Кардиология №5 / 2016
Дилатация камер сердца, вызванная мутацией гена ламина (LMNA)
1Республиканский научно-практический центр «Кардиология», Минск, Беларусь; 2Институт генетики и цитологии НАНБ, Минск, Беларусь
В статье представлена проблема дифференциальной диагностики и верификации ламин-ассоциированной дилатационной кардиомиопатии (ДКМП). ДКМП, обусловленная мутациями ядерного гена ламина (LMNA), часто связана с нарушениями ритма сердца, проводимости и различными скелетно-мышечными расстройствами. Представленный клинический случай новой ламиновой миссенс-мутации, ассоциированной с тяжелой формой ДКМП с «перекрывающимся» фенотипом аритмогенной правожелудочковой кардиомиопатии, демонстрирует лечебно-диагностические сложности и преимущества применения в такой ситуации новой классификационной системы «MOGE(S)». Важным аспектом в клинической практике кардиолога является генетическая диагностика ламиновых мутаций, позволяющая своевременно определить оптимальную тактику лечения и необходимость имплантации кардиовертера-дефибриллятора.
Дилатационная кардиомиопатия (ДКМП) — наиболее часто встречающееся среди первичных поражений миокарда заболевание, характеризующееся высокой смертностью. По данным Европейского общества кардиологов, летальность при ДКМП составляет 20% в течение 5 лет после установления диагноза. Распространенность заболевания в европейской популяции составляет более 37 случаев на 100 тыс. населения [1].
Основными причинами смерти при ДКМП являются внезапная сердечная смерть (ВСС) или смерть от декомпенсированной застойной сердечной недостаточности (СН). ДКМП может быть результатом вирусных миокардитов, системных заболеваний, токсических воздействий, метаболических и аутоиммунных расстройств, а также генетических нарушений. Заболевание отличается выраженной клинической и генетической гетерогенностью [1—3].
В настоящее время известно более 50 генов, мутации которых ассоциированы с ДКМП [4—7]. По данным разных авторов, на долю первичных ДКМП, обусловленных мутациями генов, которые кодируют структурные белки миокарда, приходится не менее 25—40% зарегистрированных случаев, по некоторым данным — до 60% [5—8].
В последнее десятилетие активно изучается ламиновый генотип ДКМП, прогностически наиболее неблагоприятный среди других генных мутаций.
Впервые мутации в гене ламина (LMNA), вызывающие развитие ДКМП с нарушением проводимости, выявлены D. Fatkin в 1999 г., затем это открытие инициировало целый ряд аналогичных исследований [9—12]. Согласно результатам мета-анализа, проведенного Van Berlo и C. Meune, у пациентов — носителей ламиновых генных аномалий, отмечается высокая летальность: в течение 3-летнего периода наблюдения зарегистрировано 46% случаев ВСС, несмотря на наличие пейсмейкеров в группе больных брадиаритмиями [11]. В исследовании М. Pasotti период наблюдения пациентов с ламин-ассоциированной ДКМП составил 36—107 (медиана 57) мес: в 55,1% случаев выявлены желудочковые тахиаритмии, в 24,5% имплантированы кардиовертерыдефибрилляторы (КД), в 32,7% зарегистрирована ВСС, а в 30,6% случаев проведена трансплантация сердца [12]. В период с 2009 по 2012 г. европейскими кардиологами (рабочие группы специалистов по проблеме) и Американской ассоциацией по сердечной недостаточности обновлены практические рекомендации по генетической диагностике кардиомиопатий (КМП) с включением специальной позиции по лечению и диагностике ламин-связанной ДКМП [13—15]. Основные положения европейских и американских кардиологов совпадают по концепции выделения ламиновых фенотипов ДКМП для обязательного молекулярно-генетического тестирования и в случае идентификации LMNA-ассоциированной ДКМП – ранней имплантации КД для профилактики ВСС [16—18].
Ламин A/C — структурный белок ядерной ламины, относящийся к классу промежуточных филаментов V. Ядерная ламина — это структурный элемент ядра, который противостоит силам деформации и защищает хроматин от физических повреждений. Ламина предопределяет размер, форму и прочность ядерной оболочки. Свойства этого элемента зависят не только от структурного строения ламинов, но и от способа его взаимодействия с компонентами цитоскелета клетки [19]. С помощью основного домена ламины формируют димер и взаимодействуют с хроматином и другими ключевыми белками внутренней ядерной мембраны, ламиновые комплексы принимают участие и в регуляции транскрипционных процессов. Снижение (потеря) функции, в частности ламина A, ведет к дегенерации, снижению нормальной регенерации клеток в различных тканях и к преждевременной клеточной гибели — апоптозу [20].
Мутации гена LMNA служат причиной 9 различных наследственных заболеваний, получивших название «ламинопатии» (в том числе мышечная дистрофия Эмери—Дрейфуса, поясно-конечностная мышечная дистрофия 1В типа, ДКМП 1А типа, болезнь Шарко—Мари—Тута типа 2В1, семейная липодистрофия Даннигана, акромандибулярная дисплазия, синдром прогерии Хатчинсон—Гилфорда). Ламинопатии отличаются значительной фенотипической и генетической гетерогенностью.
Они являются результатом миссенс-мутаций (замена аминокислоты в белке) в кодирующих частях гена ламина (72% всех изученных мутаций LMNA), в сайтах сплайсинга на границе экзон—интрон (7%), а также вставок/делеций нуклеотидов в кодирующих (9%) и некодирующих (9%) частях гена. Встречаются также нонсенс-мутации (5%), приводящие к преждевременному обрыву синтеза цепи белка — «стоп кодон» [21—24]. Практически все идентифицированные в гене LMNA мутации зарегистрированы и доступны для ознакомления в европейской базе данных UMD-LMNA мутаций (www.umd.be/LMNA) и американской базе данных (www.hgvs.org; Human Intermediate Filament Database LMNA; IPN Mutations LMNF; LMNA homepage — Leiden Muscular Dystrophy pages).
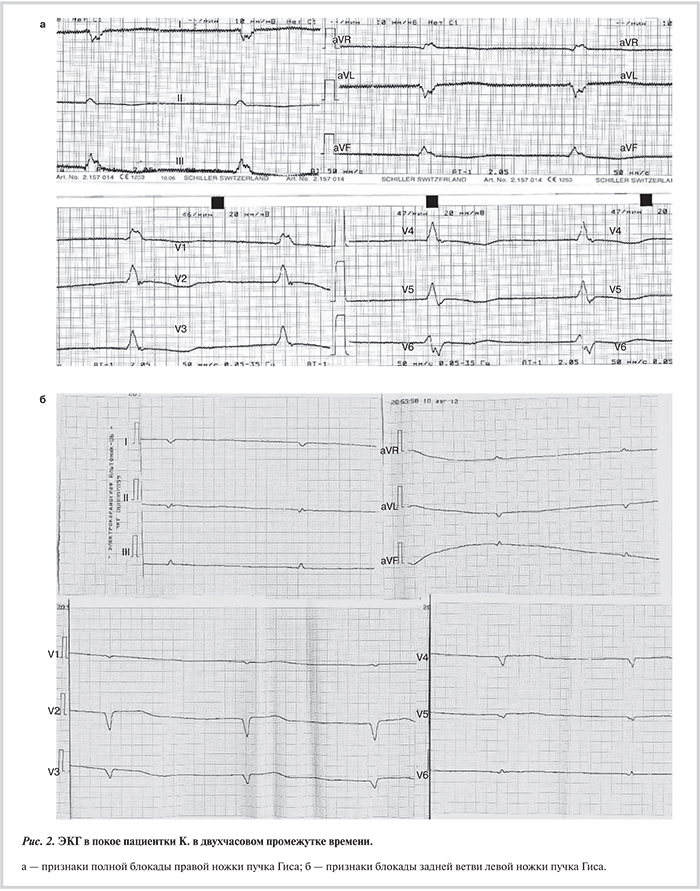
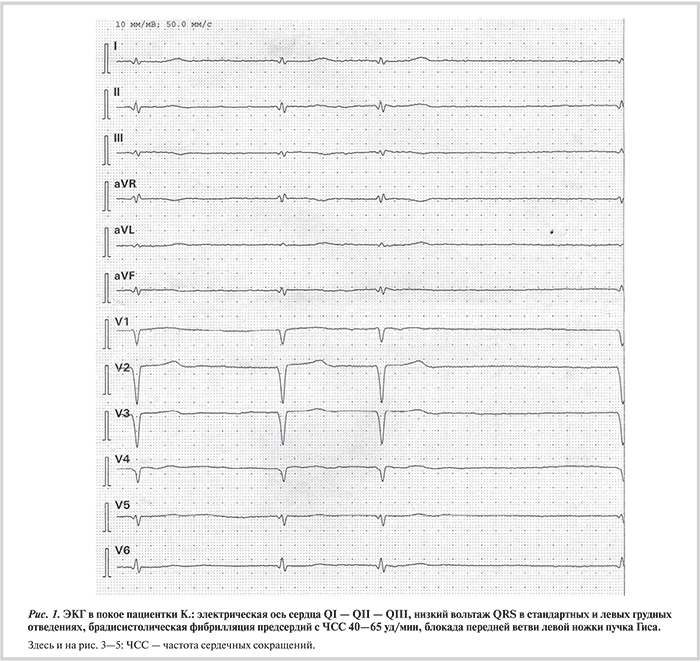
Наряду с дилатацией и снижением систолической функции левого желудочка (ЛЖ) ламиновый фенотип почти всегда сопровождается клинически значимыми нарушениями проводимости и ритма [10—13, 16—18]. Боле...












