Клиническая Нефрология №1 / 2018
Клинический случай папиллоренального синдрома
Научно-исследовательский клинический институт педиатрии им. акад. Ю.Е. Вельтищева ФГБОУ ВО РНИМУ им. Н.И. Пирогова; Москва, Россия
В статье представлено клиническое наблюдение и обзор литературы, посвященной папиллоренальному синдрому. Папиллоренальный синдром (ПРС) – редкое аутосомно-доминантное заболевание, возникающие вследствие мутации гена PAX2 (OMIM 120330). К фенотипическим особенностям наблюдаемого нами пациента с мутацией во 2-м экзоне гена РАХ2 относятся сочетание двусторонней гипоплазии почек и колобомы дисков зрительных нервов, а также не описанный ранее у этих пациентов синдром дисфункции проксимальных канальцев, проявляющийся непостоянной глюкозурией, неселективной аминоацидурией, высоким уровнем β2-микроглобулина. На примере представленного клинического случая показана необходимость комплексного подхода к обследованию детей с аномалией развития почек для ранней диагностики синдромальных вариантов патологии, что важно для определения нефрологического, витального, социального и семейного прогнозов заболевания.
Введение
В настоящее время папиллоренальный синдром (ПРС) рассматривается как первичная сосудистая дисгенезия, характеризующаеся поражением зрительного нерва и центральной нервной системы, органов мочевой системы, внутреннего уха [2]. Клинически синдром характеризуется прежде всего патологией глаз (77%) и патологией почек (92%) [3].
Аномалии глаз варьируются от бессимптомных сосудистых аномалий сетчатки до тяжелых нарушений зрительного анализатора и могут включать дисплазию диска зрительного нерва различной степени выраженности, колобому сетчатки, сосудистой оболочки и радужки [4]. Менее распространены сочетанные пороки развития глаз: стафилома склеры, аномалии желтого пятна, аномалии хрусталика, отслоение сетчатки [4]. Острота зрения колеблется от близкой к норме до серьезных нарушений на один или оба глаза (в 75%), также у пациентов может отмечаться нистагм и косоглазие [2].
Проявления со стороны почек включают гипоплазию почек, которая может быть как двусторонней, так и односторонней, мультикистоз почек, пузырно-мочеточниковый рефлюкс (ПМР). Хроническая почечная недостаточность (ХПН) может возникать в любом возрасте, при этом исход в терминальную хроническую почечную недостаточность (тХПН) отмечен почти у всех пациентов.
Примерно у 7% пациентов с ПРС имеет место нарушение слуха в виде высокочастотной сенсоневральной тугоухости; возраст начала потери слуха и его прогрессирования остается неизвестным и не оценивался проспективно [5]. В единичных случаях у пациентов имели место другие аномалии развития: пороки развития нервной и сердечно-сосудистой систем, аномалии развития половых органов, гиперэластичность кожи, слабость связочного аппарата [6].
Многообразие клинических проявлений при ПРС связано с экспрессией гена PAX2 в мезанефросе, эмбриональной щели зрительной чашки, заднем мозге, на ранних стадиях онтогенеза.
Первое упоминание о заболевании было сделано G. Rieger (1977), который сообщил о семье, в которой у отца наблюдались аномалии дисков зрительных нервов и он умер от хронического нефрита. У его сына также имелись аномалии дисков зрительных нервов и сетчатки глаза, при этом функция почек была сохранной; у дочери наблюдались аномалии развития глаз и хроническая почечная недостаточность [7].
Впервые термин ПРС предложен в 1990 г. S. Weaver и соавт. в 1988 г., которые впервые отметили и описали у 2 братьев колобому зрительного нерва и отметили связь этого симптома с заболеванием почек [8]. ПРС наследуется по аутосомно-доминантному типу и возникает в результате мутации одной копии гена PAX2, расположенного на хромосоме 10-го региона q24.3-q25.1 (рис. 1).
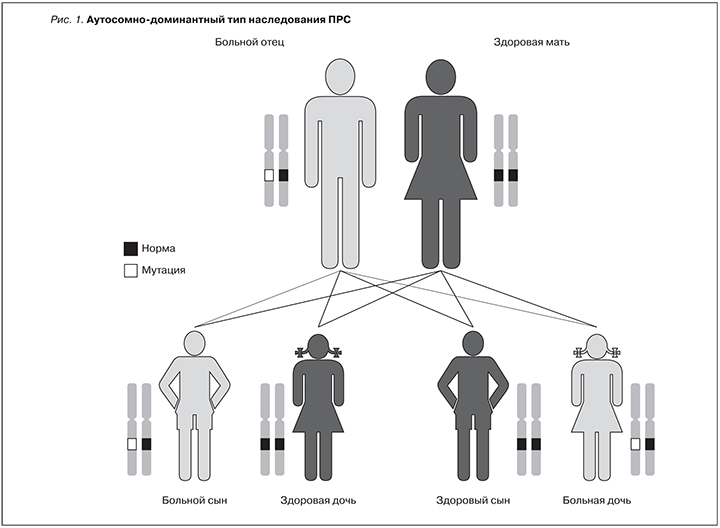
Заболевание обусловлено мутацией в гене PAX2, который кодирует фактор транскрипции парных окон ДНК-связывающих белков, участвующих в развитии урогенитального тракта, зрительного нерва и сетчатки, а также центральной нервной системы и внутреннего уха [9]. Этот ген играет важную роль в эмбриогенезе, запуская молекулярный каскад реакций во время перехода от недифференцированной мезенхимы к мезонефросу [2, 13].
 Кроме того, белок связывает отдельные участки ДНК и регулирует активность других генов [9].
Кроме того, белок связывает отдельные участки ДНК и регулирует активность других генов [9].
Роль мутации в гене PAX2 продемонстрирована в экспериментальных исследовательских работах, проводимых на биологических моделях. У трансгенных мышей была смоделирована мутация в этом гене в тех же областях, что и у пациентов с ПРС. В результате в исследуемой линии мышей обнаружены аномалии развития сосудов сетчатки и дисков зрительных нервов (рис. 2) [9].
При исследовании ткани почек у той же линии мышей продемонстрированы аномалии развития, которые проявлялись в виде двустор...












