Клиническая Нефрология №1 / 2013
Вторичный гиперпаратиреоз у больных хронической болезнью почек. Роль фактора роста фибробластов-23 (FGF-23) и Klotho
ГБOУ ВПО “Первый МГМУ им. И.М. Сеченова” Минздрава России, Москва
Проанализированы современные представления о развитии и прогрессировании вторичного гиперпаратиреоза, основанные на новых данных о патофизиологии и молекулярных механизмах взаимодействия фосфат-регулирующих систем – паратиреоидного гормона, FGF-23/Klotho и кальцитриола.
Результаты исследований, направленных на детализацию патогенеза вторичного гиперпаратиреоза при хронической болезни почек, в последние годы дополнились принципиально новыми данными. Установлены ранее не известные факторы, продуцирующиеся почечной и костной тканью, участвующие в регуляции гомеостаза фосфора, витамина D и минерализации костной ткани, – морфогенетические белки (FGF-23 и Klotho) [1, 2, 4]. В настоящее время поддержание постоянства физиологической плазменной концентрации фосфора рассматривается как результат комплексного взаимодействия паращитовидных желез (ПЩЖ), почек и костной ткани. При этом четко установлена активная эндокринная роль костной ткани [2–6].
При почечной недостаточности нарушаются все звенья минерального и костного обмена. Уже при снижении СКФ ниже 60 мл/мин/1,73 м2 уменьшается фильтрация фосфора и повышается его сывороточная концентрация, что вызывает повышение секреции FGF-23 и ПТГ; у детей аналогичные
изменения начинаются с ХБП II стадии [1, 4, 7].
Установлено, что уровень FGF-23 повышается в сыворотке крови при ХБП по мере снижения СКФ, значительно опережая увеличение сывороточной концентрации фосфора [4, 8, 9].
FGF-23 подавляют реабсорбцию фосфора, таким образом нормализуя его уровень в сыворотке крови, но при падении СКФ ниже 30 мл/мин/1,73 м2 этот механизм поддержания нормальной сывороточной концентрации фосфора становится недостаточно эффективным и развивается стойкая гиперфосфатемия, что стимулирует усиленную секрецию FGF-23 и в дальнейшем – ПТГ [1, 4, 7, 8].
Поскольку уровень сывороточного FGF-23 у больных ХБП часто предшествует гиперфосфатемии, резистентность к FGF-23 может быть одним из наиболее ранних проявлений нарушения метаболизма фосфора при ХБП [8]. Предполагают, что развитие резистентности к FGF-23 вызвано снижением
почечной экспрессии Klotho, который является кофактором действия FGF-23 [2, 4, 6, 10]. В связи с этим низкий уровень экспрессии трансмембранной формы белка Klotho в почках может быть фактором неблагоприятного отдаленного прогноза больных ХБП [2, 3, 6, 10–12].
Центральная роль FGF23/Klotho в механизмах развития ВГПТ в настоящее время является доказанной. С одной стороны, увеличение продукции FGF-23 начиная с ранних стадий ХБП препятствует развитию гиперфосфатемии и объясняет, почему сывороточная концентрация фосфора остается нормальной вплоть до выраженного снижения СКФ. С другой стороны, FGF-23 приводит к ингибированию образования кальцитриола и формированию ВГПТ за счет ослабления геномных механизмов контроля синтеза иПТГ [8, 13–15].
Получены подтверждения наличия в ПЩЖ внутриклеточных путей, общих для передачи различных внеклеточных сигналов, таких как изменения концентрации кальция, фосфора, 1,25(OH)2D3 и FGF-23/FGFR/Klotho, направленных и на регуляцию геномной экспрессии ПТГ, его секрецию, а
также на пролиферативные процессы в органе и механизмы пролиферации клеток ПЩЖ [16]. A.S. Dusso и соавт. (2001) показали, что на ранних стадиях ХБП ограничение потребления фосфора блокирует развитие гиперплазии ПЩЖ в результате специфической индукции мРНК и синтеза р21-ингибитора циклин-зависимой киназы. Напротив, диета с большим содержанием фосфора индуцировала трансформирующий фактор роста альфа (TGF-α – лиганд EGFR) и его конвертазу ADAM17 (асимметричный диметиларгинин 17), который, полагают, является аутокринным сигналом стимуляции клеточного роста и гиперплазии ПЩЖ [16–18].
Установлено, что ADAM17 может наряду с другими мембранными протеазами (BACE1, ADAM10) принимать участие в протеолизе белка Klotho [14]. При этом Klotho утрачивает свои рецепторные свойства, формируя развитие FGF-23-резистентности органов-мишеней. В то же время отщепленная
внеклеточная (экстрацеллюлярная) часть молекулы Klotho, попадая в циркуляцию, имеет не зависимые от FGF-23 плейотропные биологические эффекты в виде ингибирования Na-Р-транспортеров (NPT2a, NPT2c, NPT3), активации кальциевого и калиевого ионных каналов (TRPV5, TRPV6, ROMK1) [11,
19–21]. Активация ADAM17 связана и со снижением продукции кальцитриола при ВГПТ, поскольку VDR-активаторы блокируют его экспрессию наряду с инактивацией EGFR [22]. Следует также отметить, что активация ADAM17 в почке под действием ангиотензина II может приводить к эффектам, имеющим системное значение. ADAM17-опосредованное локальное высвобождение и увеличение в циркуляции TNF-α, обладающего известными профибротическими/провоспалительными свойствами, и молекул адгезии (ICAM-1 и VCAM-1), могут приводить к развитию системного воспалительного стресса и сердечно-сосудистых осложнений [24].
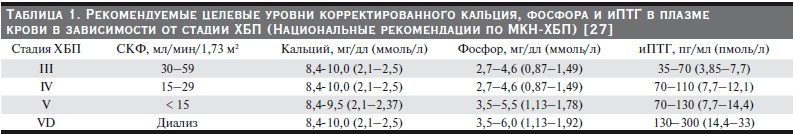
Таблица 1. Рекомендуемые целевые уровни корректированного кальция, фосфора и иПТГ в плазме
крови в зависимости от стадии ХБП (Национальные рекомендации по МКН-ХБП) [27].
Пролиферативные процессы в ПЩЖ также могут контролироваться Pit-1 – натрий-зависимым ко-транспортером фосфора, активность которого в свою очередь регулируется фосфором













{kind=link}