Фарматека №s5-17 / 2017
Всегда ли тромбоцитопения – проблема гематолога? Лечебно-диагностические алгоритмы в практике терапевта и гастроэнтеролога
ФГБОУ ВО «СЗГМУ им. И.И. Мечникова» Минздрава России, Санкт-Петербург
За последние годы получено достаточно доказательных данных о развитии тромбоцитопении при различных патологических состояниях, что указывает на необходимость знаний и опыта врачей различных клинических специальностей при проведении лечебно-диагностических мероприятий. В статье рассматриваются механизмы развития тромбоцитопении. Приводятся клинический алгоритм и лабораторно-инструментальные методы исследования, позволяющие выяснять генез тромбоцитопении. Особое внимание уделяется механизмам развития тромбоцитопении при заболеваниях пищеварительной системы.
Введение
Принято считать, что диагностика и лечение тромбоцитопении (ТП) находятся в сфере профессиональной компетенции специалиста-гематолога. Вместе с тем к настоящему моменту накоплена масса данных, свидетельствующих о снижении числа тромбоцитов при различных патологических состояниях. Это объясняет необходимость более широкого обсуждения данной проблематики среди врачей различных клинических специальностей.
Общие положения
Термин «тромбоцитопения» обычно используют при уровне тромбоцитов ниже 100,0×109/л, хотя нормальным уровнем тромбоцитов принято считать таковой в пределах от 150,0×109/л до 450,0×109/л [1]. Поэтому ряд экспертов выделяют латентную ТП при уровне тромбоцитов от 100,0×109/л до 150,0×109/л [2]. Выделение латентной ТП, на наш взгляд, обоснованно с практической точки зрения. С одной стороны, такая клиническая ситуация требует динамического наблюдения за уровнем тромбоцитов независимо от причины, с другой – при числе тромбоцитов 100,0×109/л и более полностью обеспечивается гемостаз, что, как указывается в большинстве имеющихся руководств, безопасно в контексте риска развития кровотечений [3] и что позволяет проводить различные оперативные вмешательства, в т.ч. и родоразрешение, при указанном уровне тромбоцитов.
Более того, концентрация тромбоцитов от 100×109/л до 50×109/л, протекающая без спонтанного геморрагического синдрома, может также считаться безопасной. В случаях появления признаков кровоточивости при указанном числе тромбоцитов следует искать дополнительный фактор, провоцирующий геморрагический синдром, или учитывать наличие сосудистой патологии, например, у пациентов преклонного возраста. Существующие подходы указывают, что коррекцию ТП следует проводить при числе тромбоцитов от 50×109/л до 30×109/л только при наличии геморрагических проявлений. Критическим для развития опасных для жизни геморрагических проявлений является содержание тромбоцитов ниже 10,0×109/л. Пациенты с таким уровнем ТП нуждаются в безотлагательной терапии независимо от степени клинических проявлений геморрагического синдрома [3, 4].
Этиопатогенез ТП
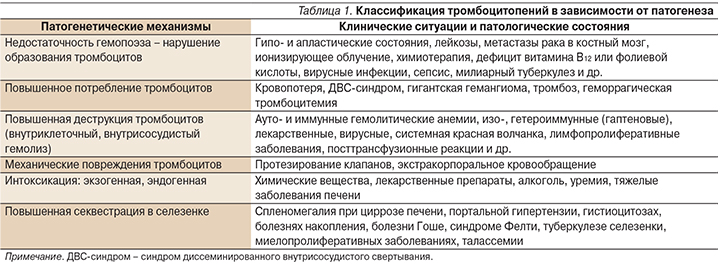
Говоря о ТП, следует подчеркнуть разнообразие приводящих к ней патогенетических механизмов и еще большее количество патологических состояний, при которых данные патогенетические механизмы реализуются [5]. Выделяют два основных механизма патогенеза ТП (табл. 1): нарушение образования тромбоцитов и повышенное их разрушение.
Диагностические алгоритмы при ТП
При выявлении у пациента ТП весьма важен детальный анализ истории заболевания, в частности установление предшествовавших развитию ТП факторов: бактериальная или вирусная инфекция, в т.ч. и вирусные гепатиты; вакцинация или применение лекарственных препаратов; пребывание в странах с риском заражения инфекционными заболеваниями (малярия, риккетсиоз, лихорадка Денге и др.); употребление алкогольных и хинин-содержащих напитков; варикозная болезнь, тромбозы, сердечно-сосудистая патология и ее терапия антикоагулянтами и дезагрегантами; наличие сопутствующих заболеваний, особенно аутоиммунных, инфекционных или опухолевых, протекающих с ТП, ДВС-синдромом; трансфузии; пересадка органов в анамнезе; беременность; наличие и длительность кровотечений после хирургических вмешательств.
В семейном анамнезе обязательно уточнить наличие болезни системы кроветворения у родственников.
При проведении объективного исследования следует активно выявлять такие симптомы, как гипо- или гипертермия, снижение массы тела, симптомы интоксикации, гепато- и спленомегалия, лимфаденопатия, патология молочных желез, сердца, вен нижних конечностей, а также врожденные аномалии. Все эти данные не специфичны.
Лабораторно-инструментальная диагностика включает несколько направлений. В клиническом анализе крови обязательны оптический подсчет числа тромбоцитов и оценка морфологии тромбоцитов (микроформы и гигантские тромбоциты), при этом следует помнить, что при наличии агрегатов тромбоцитов для исключения «ложной» ТП при использовании консерванта ЭДТА (этилендиаминтетраацетат кальция-натрия) необходим повторный анализ (используется пробирка с цитратом натрия). Всем пациентам проводится ультразвуковое исследование или компьютерная томография (КТ) органов брюшной полости и забрюшинного пространства, рентгенография или КТ органов грудной клетки, а также обследование для исключения онкологических заболеваний. Принимая во внимание, что идиопатическая тромбоцитопеническая пурпура (ИТП) – диагноз исключения, спектр используемых лабораторных тестов достаточно широк (табл. 2), по своей диагностической значимости их подразделяют на обязательные, потенциально полезные и тесты с недоказанной информативностью.

Принципы лечения ИТП
В лечении ИТП выделяют несколько линий терапии. На первом этапе используются гормоны (












