Фарматека №s1-11 / 2011
Адреногенитальный синдром: современные аспекты диагностики и лечения
Институт детской эндокринологии ФГУ ЭНЦ, Москва
Адреногенитальный синдром (АГС, врожденная дисфункция коры надпочечников – ВДКН, врожденная гиперплазия коры надпочечников – ВГКН) – группа заболеваний с аутосомно-рецессивным типом наследования, в основе которых лежит дефект одного из ферментов стероидогенеза. АГС встречается относительно часто, проявляется надпочечниковой недостаточностью, нарушением формирования пола или преждевременным половым развитием. Без соответствующей заместительной терапии заболевание представляет угрозу для жизни пациента, особенно в период новорожденности. Своевременная диагностика и лечение позволяют обеспечить пациенту и его семье хорошее качество жизни и полную социальную адаптацию. С 2006 г. в России АГС включен в программу “Национальные приоритетные проекты” и введен неонатальный скрининг.
Введение
Адреногенитальный синдром (АГС), или врожденная гиперплазия коры надпочечников (ВГКН), является одним из самых распространенных наследственных моногенных заболеваний, одновременно представляет собой вариант хронической первичной надпочечниковой недостаточности и группу патологии полового развития, а также половой дифференцировки. Кроме того, проблема АГС в стертой (неклассической) форме занимает существенное место среди причин нарушения репродуктивного здоровья (бесплодие, невынашивание беременности). Таким образом, с проблемой АГС встречаются врачи разных специальностей: неонатологи, педиатры, эндокринологи, гинекологи, генетики. Понимание основных принципов диагностики и лечения этого заболевания врачами разных специальностей является необходимым во избежание серьезных ошибок на разных этапах оказания медицинской помощи.
Клинические варианты АГС
АГС – группа заболеваний с аутосомно-рецессивным типом наследования, в основе которых лежит дефект одного из ферментов, участвующих в синтезе кортизола.
В зависимости от фермента, в гене которого имеется дефект, на сегодняшний день известно семь нозологических вариантов АГС:
• липоидная гиперплазия надпочечников (дефект StAR-протеина);
• дефицит Р450scc (20,22-десмолазы);
• дефицит 3βГСД (3β-гидроксистероиддегидрогеназы);
• дефицит CYP17 (17α-гидроксилазы/17,20-лиазы);
• дефицит CYP21 (21-гидроксилазы);
• дефицит CYP11В1 (11β-гидроксилазы);
• дефицит POR (Р450 оксидоредуктазы).
До 95 % всех случаев АГС составляет дефицит 21-гидроксилазы. Другие нозологические формы АГС встречаются редко.
Дефект 21-гидроксилазы: классификация и клинические проявления
Ген CYP21, кодирующий фермент 21-гидроксилазу, локализован на коротком плече 6-й хромосомы. Описано более пятидесяти мутаций этого гена, приводящих к синтезу фермента со степенью активности от 0 до 60 % [1].
Частота встречаемости классических вариантов дефицита 21-гидроксилазы, рассчитываемая по данным неонатального скрининга, в разных популяциях колеблется от 1 : 10 тыс. до 1 : 18 тыс. новорожденных. Чрезвычайно высокая частота выявлена в двух изолированных популяциях: у эскимосов Западной Аляски – 1 : 280, и у жителей острова Ла Руньон в Индийском океане – 1 : 2100. Частота неклассического варианта дефицита 21-гидроксилазы значительно выше – от 0,3 до 0,01 % в мировой популяции – и достигает 3,7 % среди евреев Ашкенази. По результатам неонатального скрининга, введенного в России в 2006 г., частота данной патологии в Москве составила 1 : 10 тыс. живых новорожденных.
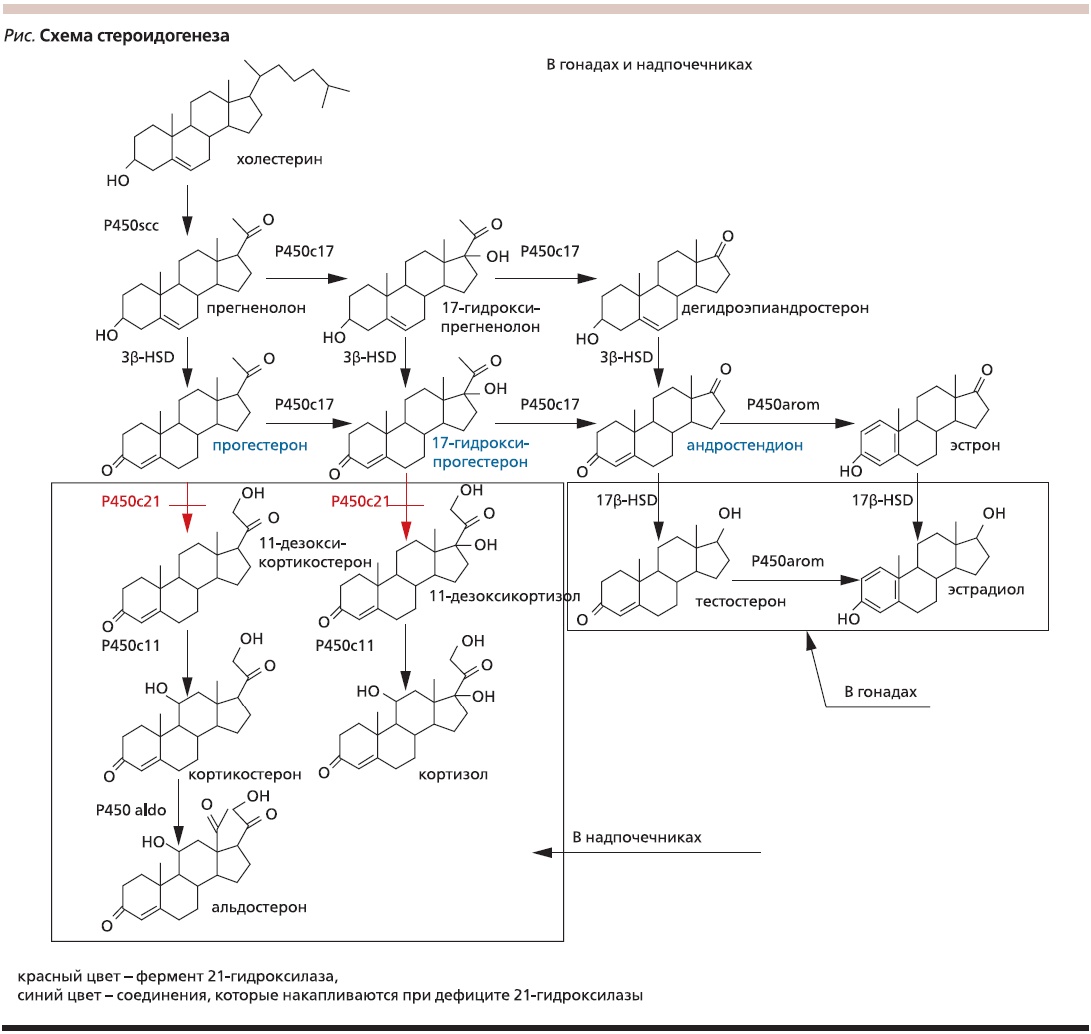
Дефект 21-гидроксилазы приводит к недостаточной продукции кортизола и ответному повышению уровня секреции адренокортикотропного гормона (АКТГ), что в свою очередь обусловливает гиперплазию коры надпочечников. Гиперплазированные надпотировать стероиды, предшествующие ферментативному блоку – прогестерон и 17-гидроксипрогестерон (17ОНП), а также андрогены, биосинтез которых не зависит от 21-гидроксилазы (см. рисунок). В результате имеется дефицит глюкокортикоидов (кортизола) и минералокортикоидов (альдостерона), а также избыток андрогенов (дегидроэпиандростерона, андростендиона, тестостерона).
Рисунок. Схема стероидогенеза.
Дефицит глюкокортикоидов (кортизола) приводит к развитию гипогликемического синдрома, что особенно тяжело проявляется у новорожденных, но также может развиваться в любом возрасте при неадекватной заместительной терапии. Развитию гипогликемии способствуют перерывы в кормлениях и интеркуррентные заболевания. Высокий уровень АКТГ, который поднимается по механизму обратной связи в ответ на низкий уровень кортизола, проявляется наличием гиперпигментаций. АКТГ – один из производных проопиемеланокортина (ПОМК)
ПОМК имеет высокое сродство к меланокортиновым рецепторам, в частности к MCR1 (рецептору меланоцитстимулирующего гормона). Большое количество АКТГ, конкурируя с меланоцитстимулирующим гормоном, связывается с его рецептором и вызывает повышение синтеза меланина в коже, что приводит к гиперпигментациям.
Дефицит минералокортикоидов (альдостерона) проявляется синдромом потери соли, который включает срыгивания, массивные рвоты “фонтаном”, полиурию, жажду, обезвоживание и низкое артериальное давление.
Избыток надпочечниковых андрогенов в эмбриональный период приводит к вирилизации наружных гениталий у плодов с кариотипом 46ХХ. Степень вирилизации наружных гениталий у девочек варьируется от 2-й до 5-й степени по шкале Прадера. После рождения избыток надпочечниковых андрогенов приводит к преждевременному половому развитию по изосексуальному типу у мальчиков и по гетеросексуальному типу у девочек. У м...













{kind=link}