Фарматека №s3-12 / 2012
Биопрепараты и прионные болезни, возможна ли этиологическая связь?
ГБОУ ВПО “Первый Московский государственный медицинский университет им. И.М. Сеченова” Минздравсоцразвития России, Москва
В обзоре литературы отражены современные данные зарубежных исследователей о природе и биологических свойствах прионных белков, об их конформационных разновидностях. Приведены сведения о роли клеточного прионного белка в функционировании различных тканей и систем организма, путях инфицирования аномальными изоформами прионов. Описаны патоморфологические изменения и клинические проявления прионных заболеваний человека, дана их современная классификация. Подробно описаны способы профилактики и предупреждения распространения прионных болезней, а также современные методы лабораторной диагностики. Дана оценка вероятности заражения в результате применения того или иного биопрепарата в зависимости от характеристики входящих в его состав агентов и метода обработки нативного материала, послужившего основой для его создания.
В последние годы отмечен рост числа биопрепаратов, для изготовления которых все шире используются различные ткани животных, в т. ч. коров и овец, т. е. животных, потенциально являющихся источником целой группы прионных заболеваний. В связи с этим актуальным становится вопрос обеспечения и оценки безопасности этих препаратов для человека. Чтобы разобраться в столь сложном вопросе, необходимо проанализировать имеющиеся сведения об этиологии, эпидемиологии, патогенезе этой группы заболеваний.
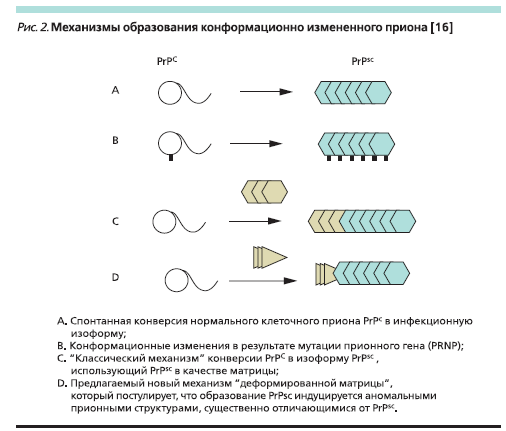
Большие успехи, достигнутые за последние 10–15 лет в области изучения прионов и вызываемых ими заболеваний, обосновали естественную потребность в систематизации накопленных данных. В табл. 1 представлена современная классификация прионных болезней человека и животных. История прионных заболеваний берет свое начало из области ветеринарии. В 1933 г. для развития каракулеводства исландские фермеры закупили в Германии большую партию овец.
Через несколько лет среди закупленных животных стали регистрироваться случаи заболевания, названного “скрепи” овец (от англ. scrappy – лоскутный), принявшего массовый характер и имевшего быстрый летальный исход. Причину этого заболевания впервые изучил доктор B. Sigurdsson. Он сфор- мулировал четыре основных признака, позволивших ему ввести новый термин для обозначения этой группы заболе ваний – “медленные инфекции”, и в1954 г. впервые прочитал цикл лекций в Лондонском университете [1].
Эти признаки и легли в основу характеристики “медленных инфекций”:
• продолжительный инкубационный период;
• медленный прогрессивный характер течения;
• необычность поражения органов и тканей;
• неизбежность смертельного исхода.
Через 3 года американский ученый D.C. Gajdusek описывает заболевание, которое встречается в горных районах острова Новая Гвинея среди папуасов-каннибалов и сейчас известно под названием “куру” [2].
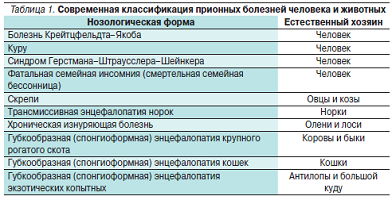
Рис. 1 Превращение нормального прионного белка в модифицированную изоформу
Рис. 2 Механизмы образования информационно измененного приона [16]
Эта болезнь характеризовалась многолетним (до 30 лет) инкубационным периодом, медленным прогрессирующим течением, поражением только головного мозга и смертельным исходом. Таким образом, проблема медленных инфекций из ветеринарии перетекла в область медицины.
Картина поражений при медленных инфекциях отличается своеобразием. Патологические изменения, обнаруживаемые в центральной нервной системе (ЦНС), характеризуются тем,что в головном, а иногда и в спинном мозге в отсутствие воспаления наблюдается гибель нервных клеток и их отростков. В тканях мозга появляются вакуоли с постепенным развитием “губкообразного” состояния. В мозговой ткани также накапливаются амилоидные бляшки, разрастается глиозная ткань. Воспаление и иммунный ответ отсутствуют. Кроме “куру” к середине 1980-х гг. стало известно еще о нескольких подобных заболеваний человека (болезнь Крейтцвельда–Якоба, синдромГерстмана–Штраусслера–Шейнкера, смертельная семейная бессонница), а также животных (трансмиссивная энцефалопатия норок, хроническая изнуряющая болезнь оленей и лосей). Подобное своеобразие патоморфоло- гических изменений в нервной ткани определило название этой группы как “трансмиссивные губкообразные энцефалопатии”. Было установлено, что возбудитель не размножается на искусственных питательных средах, проходит через бактериальные фильтры, не виден в световой микроскоп, но в то же время устойчив к ультрафиолетовому излучению, кипячению в течение 15 минут и нуклеазам. То естьего нельзя было отнести ни к вирусам, ни к бактериям, ни к вироидам. Этиология была установлена американским ученым S.B. Prusiner, который показал, что “трансмиссивные губкообразные энцефалопатии” связаны с инфицированием низкомолекулярным белком, не содержащим никаких нуклеиновых кислот. Обнаруженный белок был назван прионом [3]. За открытие прионов в 1997 г. S.B. Prusiner был удостоен Нобелевской премии.
В настоящее время принято считать, что прионы – это белковые инфекционные частицы, возбудители прионных “конформационных” болезней, которые развиваются в результате неправильного сворачивания (нарушения правильной конформации) клеточного белка, необходимого для нормального функционирования организма. Название произошло от английского словосочетания proteinaceous infectious particles – белковые инфекционныечастицы. Следует особо отметить, что прионовый протеин PrPc(cellular prion protein) – нормальная изоформа прионного белка с молекулярной массой 33–35 кД, детерминируемая геном прионного белка (PrNP), расположенного на 20-й хромосоме человека, по своей природе является сиалогликопротеином и синтезируется главным образом в нейронах, хотя продуцировать его могут и многие другие клетки. [4, 5], причем этот нормальный PrPcлокализован на поверхности клетки. Он как бы “заякорен” в богатую холестеролом мембрану клетки через молекулу гликопротеина [6] и его отличительной особенностью является чувствительность к протеазе. PrPcподдерживает качество миелиновой оболочки, регулирует перед...













{kind=link}
{kind=link}