Кардиология №5 / 2011
Диагностика, стратификация риска внезапной смерти и лечение основных молекулярно-генетических вариантов синдрома удлиненного интервала QT
ФГУ Московский научно-исследовательский Институт педиатрии и детской хирургии Минздравсоцразвития России, Детский научно-практический центр нарушений сердечного ритма, 125412, Москва, ул. Талдомская, д. 2; ФГУ Российский кардиологический научно-производственный комплекс Минздравсоцразвития России, Институт клинической кардиологии им. А.Л. Мясникова, Отдел клинической электрофизиологии и рентгенхирургических методов лечения нарушений ритма сердца
Наследственный синдром удлиненного интервала QT (СУИQT) относится к первичным электрическим заболеваниям сердца и характеризуется удлинением интервала QT на электрокардиограмме (ЭКГ) покоя, приступами потери сознания вследствие угрожающих жизни желудочковых аритмий. В обзоре обсуждаются вопросы диагностики, стратификации риска внезапной сердечной смерти и лечения пациентов с основными молекулярно-генетическими вариантами СУИQT — LQT1, LQT2 и LQT3, на долю которых приходится почти 90% всех генетически подтвержденных случаев. Представлены современные данные о связи между выраженностью фенотипических проявлений СУИQT и генетическими вариантами синдрома. Сформулированы наиболее характерные клинические и электрокардиографические дифференциально-диагностические критерии LQT1, LQT2 и LQT3, которые оцениваются на основании данных стандартной ЭКГ, поверхностного картирования ЭКГ, стресс-теста и позволяют в большинстве случаев еще до получения результатов молекулярно-генетического исследования принять решение о патогенетической терапии. Лечение больных с СУИQT заключается в исключении триггеров угрожающих жизни аритмий, характерных для каждого варианта синдрома и специфических для каждого пациента; исключении препаратов, способных удлинять интервал QT, и в систематическом применении β-адреноблокаторов, которые остаются основным средством медикаментозной антиаритмической терапии. В качестве вторичной профилактики внезапной сердечной смерти и при недостаточной эффективности антиаритмической терапии показана имплантация кардиовертера-дефибриллятора.
В течение последних двух десятилетий значительный прогресс в понимании электрофизиологических основ внезапной аритмической смерти во многом был обусловлен интенсивными исследованиями синдрома удлиненного интервала QT (СУИQT), при котором имеется высокий риск внезапной сердечной смерти (ВСС) вследствие развития угрожающих жизни желудочковых аритмий. Данный синдром относится к первичным электрическим заболеваниям сердца [1] и характеризуется удлинением интервала QT на электрокардиограмме (ЭКГ) покоя, приступами потери сознания вследствие полиморфной желудочковой тахикардии (ЖТ), тахикардии типа «пируэт» или фибрилляции желудочков [2]. С момента открытия синдрома до настоящего времени это заболевание служит уникальной моделью для изучения патофизиологии, электрокардиографических проявлений, молекулярно-генетических механизмов, а также факторов риска внезапной аритмической смерти. Именно исследования СУИQT позволили понять механизмы так называемых первичных электрических заболеваний сердца, или каналопатий, проявляющихся злокачественными нарушениями ритма сердца (НРС) с высоким риском ВСС в отсутствие органических изменений сердечно-сосудистой системы. В основе этих заболеваний лежат мутации в генах, ответственных преимущественно за нарушение функций ионных каналов. Созданы и постоянно ведутся международные и локальные регистры случаев заболевания [3]. Существенно изменились взгляды на этиологию и патогенез синдрома — первоначальная теория сцепленного с полом наследования [4] уступила место представлениям о фенотипической и генотипической разнородности с дифференцированными подходами к лечению и принципиально отличающимся прогнозом при различных формах заболевания [2, 5]. Дальнейшее изучение молекулярно-генетических основ синдрома направлено на выявление новых мутаций, определение вклада генов-модификаторов и сочетанных мутаций в патогенез и прогноз заболевания. По возможности ранняя, доклиническая диагностика СУИQT и лечение с учетом молекулярно-генетического варианта синдрома, индивидуальных факторов и маркеров риска позволяют в большинстве случаев предотвратить ВСС, значительно повысить продолжительность и качество жизни больных.
История открытия синдрома
В 1957 г. A. Jervell и F. Lange-Nielsen [6] сообщили о норвежской семье, в которой 4 из 6 детей страдали врожденной тугоухостью и частыми приступами потери сознания, провоцируемыми эмоциями или физической нагрузкой, и имели удлинение интервала QT на ЭКГ покоя (табл. 1). В 1963 г. C. Romano [7] и в 1964 г. O.C. Ward [8] описали семьи, в которых удлинение интервала QT было выявлено у одного из родителей и нескольких детей без глухоты. Впоследствии было подтверждено существование 2 форм — синдрома Джервелла—Ланге-Нильсена (аутосомно-рецессивный тип наследования) и Романо—Уорда (аутосомно-доминантный тип наследования), получивших название по фамилии авторов, впервые их описавших [9].
Таблица 1. История изучения СУИQT
Примечание. Здесь и в табл. 2: СУИQT — синдром удлиненного интервала QT.
Наследственная природа синдрома была подтверждена методами молекулярно-генетического анализа только в начале 90-х годов ХХ века, когда М. Keating и соавт. выявили ген KCNQ1, ответственный за развитие наиболее распространенного молекулярно-генетического варианта синдрома (LQT1) [10]. В последующем были определены 12 генетических вариантов СУИQT (см. табл. 1; табл. 2). За развитие клинических проявлений СУИQT ответственны не менее 11 генов, получивших числовую нумерацию (вариант) согласно хронологии их открытия (LQT1—LQT12) [11]. Мутации идентифицируются у 50—70% больных с клинически установленным диагнозом [2], что предполагает существование и других неизвестных пока генов, связанных с этим синдромом.
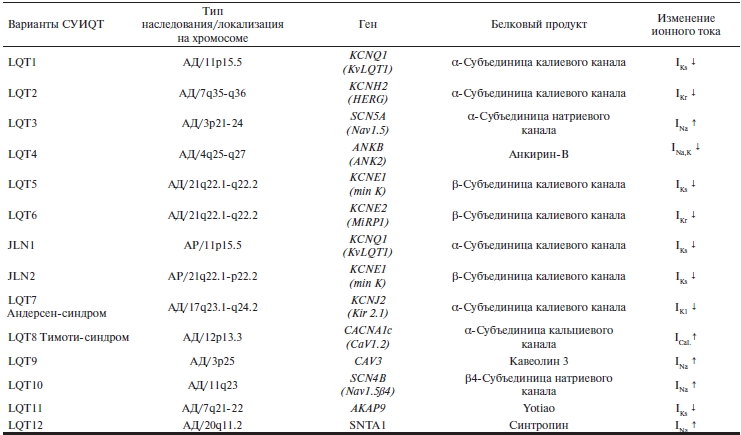
Таблица 2. Молекулярно-генетические варианты СУИQT [11]
Примечание. АД — аутосомно-доминантный; АР — аутосомно-рецессивный; LQT — синдром удлиненного интервала QT; JLN — синдром Джервелла—Ланге-Нильсена; IKs — медленный калиевый ток задержанного выпрямления; IKr — быстрый калиевый ток задержанного выпрямления; IK1 — активируемый кальцием калиевый ток; INa — натриевый ток; INa,K — натриево-калиевый ток; ICaL — медленный кальциевый ток; ↓ — снижение; ↑ — повышение.
Синдром выявляется в европейской и американской популяциях с частотой до 1:2500—1:7000 при пенетрантности 90% [2, 12]. При этом распространенность молекулярно-генетических вариантов синдрома Романо—Уорда различна. Описаны семьи со спорадическими случаями СУИQT и очень низкой пенетрантностью, составляющие около 25% (LQT4—LQT12). Большинство пациентов с установленным молекулярно-генетическим диагнозом относятся к первым 3 вариантам синдрома, соответственно LQT1 выявляется в 50—55% ...













{kind=link}
{kind=link}