Кардиология №3 / 2014
Дилатационная кардиомиопатия, вызываемая мутацией p.E446K в гене SCN5A
ФГБУ Российский научный центр хирургии им. акад. Б.В. Петровского РАМН, 119991 Москва, Абрикосовский пер., 2; ГБОУ ВПО Первый МГМУ им. И.М. Сеченова Минздрава России; ФГБУ Центральная клиническая больница с поликлиникой Управления делами Президента Российской Федерации
Дилатационная кардиомиопатия (ДКМП) — заболевание миокарда, характеризующееся прогрессирующим увеличением размеров камер сердца и снижением сократительной способности миокарда; оно является наиболее частой причиной тяжелой сердечной недостаточности, при которой требуется трансплантация сердца. Частота развития заболевания в популяции составляет около 36,5:100 000 населения. В 20—30% случаев ДКМП имеет семейный характер. Первичная ДКМП — генетически гетерогенное состояние, которое могут вызывать мутации более чем в 30 разных генах. В 2004 г. впервые выявлена роль мутаций в гене SCN5A как причина ДКМП, сопровождающейся нарушениями ритма и проводимости. В данной работе мы представляем клинический случай семейной ДКМП с нарушением ритма и проводимости, вызванной мутацией p.E446K в гене SCN5A. В 2 поколениях наблюдались ДКМП с умеренной гипертрофией миокарда, прогрессирующее нарушение атриовентрикулярной проводимости, фибрилляция предсердий и врожденный порок сердца (дефект межпредсердной перегородки), при этом порок сердца наследовался независимо от ДКМП и нарушений ритма сердца и не сегрегировал с наличием мутации.
Дилатационная кардиомиопатия (ДКМП) — заболевание миокарда, характеризующееся прогрессирующим увеличением размеров камер сердца и снижением сократительной способности миокарда; оно является наиболее частой причиной тяжелой сердечной недостаточности, при которой требуется трансплантация сердца [1]. Частота развития заболевания в популяции составляет около 36,5:100 000 населения, однако даже эти данные могут быть заниженными [2].
В 20—30% случаев ДКМП имеет семейный характер [3]. Заболевание генетически гетерогенное, в настоящее время известны более 100 генов, мутации в которых могут быть причиной кардиомиопатии (КМП) [4]. Поэтому ДНК-диагностика этого заболевания остается исключительно сложной задачей, а данные о клинических особенностях различных генетических форм заболевания весьма ограничены. В 2004 г. впервые показана роль мутаций в гене SCN5A как причина ДКМП, сопровождающейся нарушениями ритма и проводимости [5]. В международном каталоге наследственных заболеваний OMIM этот генетический вариант КМП обозначен ДКМП, тип 1Е, номер заболевания MIM:*601154 [6]. Этот ген кодирует α-субъединицу натриевого канала Nav1.5, ответственного за основной деполяризующий компонент потенциала действия кардиомиоцитов (КМЦ), входящий натриевый ток INa [7]. Натриевый входящий деполяризующий ток служит пусковым механизмом развития потенциала действия в КМЦ, результирующий деполяризующий ток — сигналом, инициирующим сокращение саркомерных белков отдельного КМЦ. Благодаря системе межклеточных контактов КМЦ и клеток проводящей системы сердца эти периодические деполяризации лежат в основе синхронного и ритмичного сокращения камер сердца [7].
Мутации в гене SCN5A являются причиной различных нарушений ритма сердца, таких как синдром удлиненного интервала QT, синдром Бругада, синдром слабости синусного узла, прогрессирующего нарушения проводимости [7].
Частота опосредованных SCN5A случаев в общей группе больных с ДКМП относительно невелика — около 3% [8]. Однако среди больных, у которых ДКМП сочетается с прогрессирующим поражением проводящей системы и наджелудочковыми и/или желудочковыми нарушениями ритма, их доля значительно выше. Согласно рекомендациям HRS/ESHRA (Heart Rhythm Assocoation/European Society of Heart Rhythm), больным с сочетанием предположительно первичной ДКМП и нарушениями проводимости (атриовентрикулярная — АВ-блокада любой степени) расширенное или выборочное (гены SCN5A и LMNA) генетическое исследование является рекомендованным [9].
Материал и методы
Проведено клиническое и генетическое обследование членов семьи DCM_83. Клинико-генеалогический анализ заключался в сборе и анализе семейных данных, в том числе архивного материала за период с 1992 по 2013 гг. Клиническое и инструментальное обследование включало детальный осмотр, электрокардиографию, суточное холтеровское мониторирование, ультразвуковое исследование (УЗИ) сердца и брюшной полости, мультиспиральную компьютерную томографию сердца (МСКТ), оценку функции внешнего дыхания. Лабораторные исследования включали общий и биохимический анализ крови, коагулограмму, исследование уровня гормонов щитовидной железы, оценку функции почек, ПЦР кардиотропных вирусов в крови. Получено информированное согласие от членов семьи на генетическое исследование, которое включало прямое секвенирование по Сенгеру кодирующей последовательности и прилегающих интронных областей гена SCN5A у пробанда, а также секвенирование экзона 10 (направленный поиск мутации) у членов семьи.
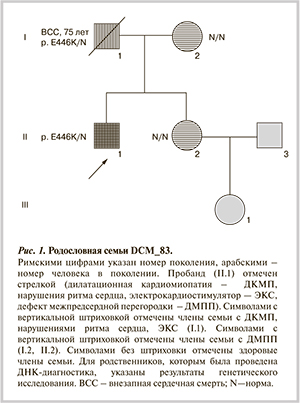 Клиническое наблюдение. В отделение хирургического лечения дисфункции миокарда ФГБУ Р...
Клиническое наблюдение. В отделение хирургического лечения дисфункции миокарда ФГБУ Р...












