Фарматека №11 (304) / 2015
Клинико-электроэнцефалографические, нейровизуализационные характеристики и эффективность антиэпилептической терапии синдрома Веста у пациентов с синдромом Дауна (результаты собственных наблюдений)
(1) Институт детской неврологии и эпилепсии им. Святителя Луки, Москва; (2) Кафедра неврологии, нейрохирургии и медицинской генетики педиатрического факультета ГБОУ ВПО РНИМУ им. Н.И. Пирогова Минздрава России, Москва
Эпилепсия у пациентов с синдромов Дауна выявляется в 8–9% случаев, при этом половину всех случаев эпилепсии составляет синдром Веста. Обследованы 8 пациентов с синдромом Дауна и синдромом Веста. Средний дебют эпилептических спазмов произошел в возрасте 5,6 месяца. Характер приступов (эпилептических спазмов), картина на электроэнцефалограмме (ЭЭГ) (гипсаритмия, или модифицированная гипсаритмия), ЭЭГ-паттерн пароксизмов в целом не отличаются от симптоматики синдрома Веста другой этиологии. Антиэпилептическая терапия привела к значительному улучшению в 87,5% случаев синдрома Веста при синдроме Дауна, при этом ремиссия достигнута у 50% пациентов. В данном исследовании были использованы тетракозактид, препараты вальпроевой кислоты, топирамат, ламотриджин, этосуксимид, вигабатрин. Показана высокая эффективность комбинации вальпроатов с топироматом.
Введение
Синдром Дауна (СД) – хромосомный синдром, характеризующийся особым своеобразным фенотипом (близко расположенные глаза с монголоидным разрезом и эпикантом; большой, не помещающийся во рту язык; характерная форма носа), умственной отсталостью в сочетании с добродушным поведением, задержкой моторного развития, гипотонией, короткими конечностями (Бадалян Л.О., 1980; Кеннет Л. Джонс, 2011) [1, 2]. Частота встречаемости составляет, по данным разных авторов, от 1:800 до 1:1000 среди здоровых детей. По данным Л.О. Бадаляна и соавт. (1971), среди детей с психомоторными нарушениями частота встречаемости СД достигает 10% [1]. При СД отмечается экстракопирование генетического материала на 21-й хромосоме [1, 3].
Выделяют несколько вариантов, приводящих к развитию данного синдрома: трисомия 21-й хромосомы, транслокация, мозаицизм. Наиболее часто (более чем в 94 случаев) у пациентов с СД отмечается трисомия 21-й хромосомы с кариотипом 47-й. Транслокационный синдром (45,XX, t(14;21q)) занимает второе место по частоте и встречается в 1,2–4,0% случаев. Мозаицизм (46,XX/47,XX,+21) выявляется в 1,0–2,4% случаев [2, 4].
Эпилепсия у пациентов с СД, по данным разных авторов, может выявляться в 8–9% случаев [2, 5, 6]. Среди больных СД, страдающих эпилепсией, синдром Веста (СВ) встречается в 49–56% случаев [4, 7]. При этом, по данным Kurokawa и соавт. (1980), среди больных СВ пациенты с СД составляют всего 1% случаев [8]. Вместе с тем Stafstrom, Konkol (1994) отмечают, что эпилептические спазмы (ЭС) при СД встречаются в 8–10 раз чаще, чем в общей популяции [9].
СВ – возраст-зависимый полиэтиологический эпилептический синдром, относящийся к группе младенческих эпилептических энцефалопатий.
СВ характеризуется следующими критериями [10, 11]:
- Особый тип эпилептических приступов – эпилептические (инфантильные) спазмы, представляющие массивные миоклонические и (или) тонические, про- и (или) ретропульсивные, симметричные и (или) асимметричные, серийные и (или) изолированные спазмы аксиальной и конечностной мускулатуры.
- Изменения на электроэнцефалограмме в виде гипсаритмии.
- Задержка психомоторного развития.
По этиологии, особенностям течения и прогнозу у пациентов с трисомией по 21-й хромосоме выделяют идиопатический и криптогенный/симптоматический варианты СВ. Согласно Silva и соавт. (1996), основные диагностические критерии идиопатического варианта СВ у пациентов с СД: отсутствие врожденных кардиопатий и признаков последствия перинатальной гипоксически ишемической энцефалопатии [12]. Для идиопатического варианта СВ при СД характерны симметричные спазмы. В единичных случаях спазмы могут быть асимметричными. При идиопатическом варианте СВ на электроэнцефалограмме (ЭЭГ) регистрируется классическая симметричная гипсаритмия [13].
По данным Stafstrom, Konkol (1994), наиболее частой причиной возникновения симптоматического варианта СВ у пациентов с трисомией 21-й хромосомы является последствие перинатальной гипоксически-ишемической энцефалопатии [9]. Escofet и соавт. (1995), Smigielska-Kuzia и соавт. (2009) отмечают важность выявления при симптоматическом варианте СВ латерализационных признаков, которые могут указывать на структурное локальное повреждение головного мозга. К этим признакам можно отнести асимметричные тонические спазмы в виде формирования «позы фехтовальщика», отведения головы и глаз в сторону. Нередко при симптоматическом варианте СВ отмечаются фокальные приступы, версивные или тонические. На ЭЭГ при симптоматическом варианте, как правило, регистрируется «модифицированная» гипсаритмия с преобладанием региональных или мультирегиональных эпилептиформных нарушений [14, 15].
Вне зависимости от этиологии после дебюта ЭС у большинства пациентов с СД наблюдается выраженная задержка или регресс психомоторного развития. При видео-ЭЭГ-мониторинге ЭС при СД характеризуются классическим ЭЭГ-паттерном: появлением высокоамплитудной билатерально-синхронной диффузной дельта-волны, которая «прерывает» картину гипсаритмии, с присоединением низкоамплитудной быстроволновой активности (lafa), сменяемой или сопровождаемой коротким эпизодом уплощения биоэлектрической активности; далее постепенно наблюдается восстановление картины гипсаритмии [12].
В отечественной литературе сочетание СД и СВ описывается в единичных публикациях [16–18]. С целью изучения анамнестических, клинических, электроэнцефалографических и нейровизуализационных особенностей у пациентов СВ и СД было проведено настоящее исследование.
Материал и методы
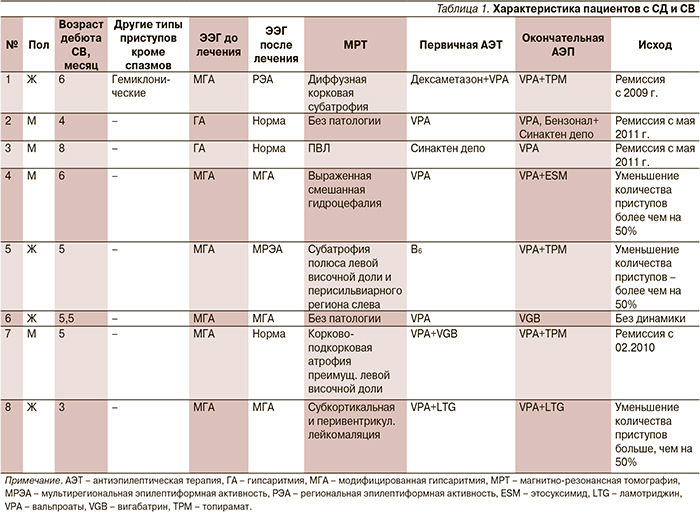
В исследование были включены 8 пациентов с генетически верифицированным СД и СВ, которые проходили обследование и лечение на клинической базе Института детской неврологии и эпилепсии им. Святителя Луки с 2006 по 2015 г.
Диагностика СВ проведена согласно критериям международной классификации эпилепсий, эпилептических синдромов и схожих заболеваний (1989), а та...












