Фарматека №3 (197) / 2010
Клинико-генетические характеристики синдрома множественных эндокринных неоплазий типа 1 и принципы его лечения
Представлены современные представления об эпидемиологии, патогенезе и клинической картине синдрома множественных эндокринных неоплазий типа 1 (МЭН-1). МЭН-1 – это редкий врожденный аутосомно-доминантный синдром, вызванный мутацией гена MEN1, локализованного на хромосоме 11q13. Для МЭН-1 характерно сочетание опухолей околощитовидных желез, гипофиза и поджелудочной железы. Рассматривается алгоритм диагностики синдрома МЭН-1, обсуждаются возможность его лечения, в т. ч. с использованием аналогов соматостатина. Подчеркивается, что на основе точного диагноза и современных методов хирургического и медикаментозного лечения пациентов с МЭН-1 в последние годы значительно улучшился прогноз и увеличилась продолжительность жизни пациентов с разнообразными проявлениями этого синдрома.
Синдром множественных эндокринных неоплазий (МЭН) – это группа генетически разнородных наследственных заболеваний, при которых выявляются опухоли или аденоматозные гиперплазии в двух и более эндокринных органах. Как правило, это нейроэндокринные опухоли (НЭО) – новообразования, происходящие из клеток APUD-системы. Эти клетки способны продуцировать нейротрансмиттеры, нейромодуляторы или нейропептиды, которые выделяются из секреторных гранул в ответ на внешние стимулы, а при опухолевой трансформации клеток их секреция становится нерегулируемой. К клеткам APUD-системы относятся островковые клетки поджелудочной железы (ПЖ), нейроэндокринные клетки респираторной системы и желудочно-кишечного тракта (ЖКТ), парафолликулярные клетки щитовидной железы (ЩЖ).
В зависимости от эмбриологического происхождения выделяют следующие группы клеток, относящихся к APUD-системе:
1. Эндокринные клетки нейроэктодермального происхождения, секретирующие серотонин, мелатонин, катехоламины.
2. Производные нервного гребешка, секретирующие адреналин, норадреналин, серотонин, кальцитонин.
3. Производные эндо- или мезодермы – гастроэнтеропанкреатические и эндокринные клетки респираторного и урогенитального трактов (G-, A-, D-, ES-, ECL-, S-эндокринные клетки). НЭО – гетерогенная группа различных по локализации, характеру роста и клинической симптоматике опухолей, происходящих из нейроэндокринных клеток и, соответственно, имеющих сходные цитологические характеристики.
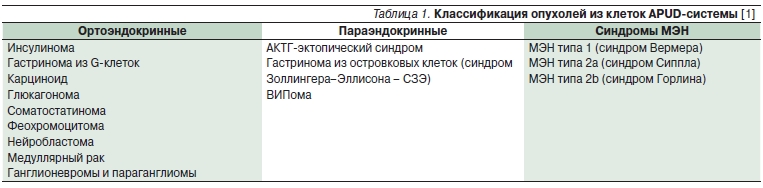
Таблица 1. Классификация опухолей из клеток APUD-системы [1].
Таблица 2. Характеристика синдромов МЭН [2, 3, 4].
Как видно из табл.1, синдромы МЭН входят в состав НЭО. Различают синдромы МЭН типов 1 и 2 (табл. 2).
МЭН-1 – редкий врожденный аутосомно-доминантный синдром, вызванный мутацией гена MEN1, локализованного на хромосоме 11q13 [5, 6]. Ген MEN1 имеет 10 экзонов (первый экзон некодирующий) и кодирует 610-аминокислотный белок менин. Хотя функции менина до сих пор полностью не изучены, предполагается его участие в репликации и репарации ДНК, транскрипции и модификации хроматина. В основном менин ведет себя как супрессор опухолевого роста [7–9], широко экспрессируемый в клетках эндокринных и неэндокринных тканей [10, 11]. У 40 % пациентов имеется сочетание опухолей ОЩЖ, гипофиза и ПЖ. Однако не у всех больных развиваются все типы опухолей. Диагноз синдрома МЭН-1 устанавливается при наличии опухолей минимум двух вышеперечисленных эндокринных желез, поражение которых наиболее характерно для синдрома МЭН-1, или при обнаружении хотя бы одной подобной опухоли у родственника пациента с синдромом МЭН-1 [11]. По данным Human gene mutation database (http://www.hgmd.cf.ac.uk/ac/index.php), в настоящее время описано более 540 различных геномных мутаций гена MEN1. Большинство из них является инактивирующим, приводящим к прекращению синтеза или изменению структуры менина и туморогенезу [12, 13]. За исключением группы геномных мутаций гена MEN1, найденных у пациентов с семейным изолированным первичным гиперпаратиреозом (ПГПТ), и мутаций MEN1-Burin в семьях с высокой частотой пролактином и низкой частотой гастрином, других генотип-фенотипических корреляций для синдрома МЭН-1 отмечено не было [12–16].
Клиническая характеристика синдрома МЭН-1 очень разнообразна даже у пациентов внутри одной семьи как по сочетанию пораженных органов, так и по возрасту начала заболевания, а также тяжести проявлений. В разных публикациях приводятся различные варианты частоты встречаемости тех или иных сочетаний поражения эндокринных желез и других органов при МЭН-1, что иногда зависит и от медицинского профиля научного учреждения, публикующего результаты своего исследования. Так, в нашем центре, который широко занимается проблемами опухолей гипофиза и ПГПТ, у 42 пациентов было выявлено сочетание ПГПТ с опухолями других эндокринных желез, наиболее часто – аденомами гипофиза (71 %). У 40 % больных имелись опухоли ПЖ, дополнительно у 30 % пациентов выявлены опухоли надпочечников. Опухоли другой локализации встречались намного реже.
У 17 пациентов на момент обследования было поражено более двух эндокринных желез одновременно, в т. ч. у 4 пациентов ПГПТ сочетался с опухолями и гипофиза, и ПЖ, и надпочечников.
По данным литературы (табл. 2) и результатам наших исследований, ПГПТ является наиболее ранним и частым клиническим проявлением синдрома с пенетрантностью до 100 % после 50 лет [17].
Энтеропанкреатические опухоли выявляются приблизительно у 60 % пациентов, причем на аутопсии эти опухоли встречаются в 100 % случаев [18]. В отличие от спорадических опухолей ПЖ МЭН-ассоциированные опухоли этого органа развиваются рано, почти всегда они мультифокальны и наблюдаются на всем протяжении железы. Клинические симптомы зависят от гормональной активности опухолей.
При гастриномах развивается СЗЭ, характеризующийся избыточной продукцией гастрина и гиперпродукцией соляной кислоты, что проявляется болями в животе, эзофагитом, пепсическими язвами и диареей [11]. Около четверти всех гастрином прих...













{kind=link}
{kind=link}