Фарматека №14 (367) / 2018
Особенности семейной формы болезни Альцгеймера с ранним началом
1) Первый Московский государственный университет им. И.М. Сеченова (Сеченовский Университет), Москва, Россия;
2) Международное общество «Стресс под контролем», Москва, Россия;
3) НМИЦ нейрохирургии им. Н.Н. Бурденко, Москва, Россия;
4) Национальный медицинский исследовательский центр профилактической медицины, Москва, Россия;
5) ПКБ № 4 им. П.Б. Ганнушкина ДЗМ, Москва, Россия
Болезнь Альцгеймера (БА) – наиболее распространенная форма деменции в клинической практике. Хуже диагностируется ранняя форма БА в связи с редкой встречаемостью. В данной статье представлен актуальный обзор данных о частоте встречаемости, патоморфологии головного мозга, клинической картине, диагностике и медикаментозном лечении БА с ранним началом. Особое внимание уделено генетическим аспектам БА с аутосомно-доминантным типом наследования.
Введение
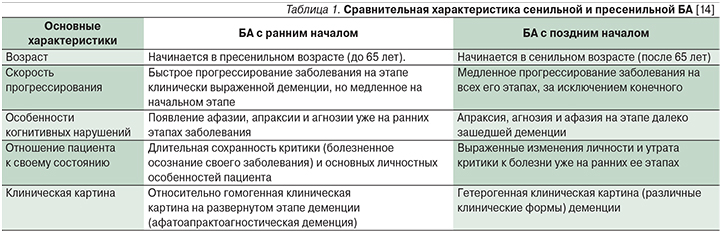
Болезнь Альцгеймера (БА) – нейродегенеративное заболевание, проявляющееся прогрессивным снижением памяти и других когнитивных функций вследствие гибели нейронов коры больших полушарий [1]. БА с ранним началом (до 65 лет) составляет 1–5% от всех случаев БА [2]. В США насчитывают примерно 200 тыс. пациентов с ранней формой БА, что как раз составляет около 4% от 5,7 млн больных БА [3]. В российских исследованиях данные о заболеваемости пациентов с БА с ранним началом отсутствуют. По данным Росстата, смертность от БА с ранним началом за 2012 г. у пациентов в возрасте 35–44, 45–54 и 55–64 года составила 0,005, 0,038 и 0,330 на 100 тыс. населения соответственно [4]. Актуальность проблемы заключается не только в том, что ранняя форма хуже диагностируется в связи с редкой встречаемостью, протекает более быстро и агрессивно, а также длительно осознается пациентом по сравнению с БА с поздним началом (табл. 1), но и в том, что пациентами становятся лица трудоспособного возраста, что способствует формированию разнообразных психологических, социальных и экономических проблем [5].
В настоящее время понятие БА с ранним началом расширено и дополнено. БА с ранним началом может иметь как семейную, так и спорадическую формы [6]. Причем около половины случаев семейной формы БА с ранним началом приходится на аутосомно-доминантную форму [7]. Семейная форма БА может относиться к заболеванию не только с ранним, но и с поздним началом (после 65 лет). Причем часть случаев семейной формы БА с поздним началом приходится также на аутосомно-доминантную форму [8]. Таким образом, существует несколько сходных, в определенной степени перекрывающихся состояний: семейная форма БА (FAD – Familial Alzheimer’s Disease) [9], БА с ранним началом (EOAD – Early-Onset Alzheimer’s Disease) [10], семейная форма БА с ранним началом (EOFAD – Early-onset familial Alzheimer’s disease) [11], аутосомно-доминантная БА (ADAD – autosomal dominant Alzheimer’s disease) [12] и аутосомно-доминантная форма БА с ранним началом (ADEOAD – early-onset autosomal dominant Alzheimer’s disease) [13].
В настоящей статье речь пойдет о семейной форме БА с ранним началом, включая аутосомно-доминантную форму.
БА с ранним началом обычно начинается в возрасте от 35 до 65 лет. Средний возраст больных составляет 54–56 лет, средняя продолжительность жизни после установления диагноза – 8–10 [15]. Заболевание сопровождается прогрессирующим нарушением памяти, интеллектуальной деятельности и высших корковых функций, приводит к развитию тотальной деменции с синдромами афазии, апраксии и агнозии [16]. Возможно проявление зрительной дисфункции и дискалькулии [17].
Также, по данным И.В. Колыхалова (2016), у 60% пациентов на том или ином этапе развития заболевания имеют место поведенческие и психопатологические расстройства следующих групп:
- Психотические расстройства (бредовые – 31%, зрительные и слуховые галлюцинации – 13 и 10% соответственно);
- Аффективные расстройства (депрессия – более 50%, тревога – от 24 до 65%, апатия – от 19 до 76%);
- Поведенческие нарушения (агрессия – 6%, двигательное беспокойство [аберрантное моторное поведение] – 12–84%) [18].
Патоморфологическая картина характеризуется церебральной атрофией с уменьшением объема и массы мозга, атрофией извилин коры, c расширением корковых борозд и желудочковой системы. При микроскопическом исследовании отмечается массивная утрата нейронов коры мозга, гиппокампа, а также базального ядра Мейнерта и голубого пятна ствола мозга. У сохранившихся нейронов выявляется дегенерация дендритов [19].
В связи с тем что семейная форма БА может дебютировать в разное время (быть БА с ранним или поздним началом), исследователи уделяют большое внимание этиологическим факторам развития заболевания. Считается, что чем позже наступает БА, тем больше доминируют факторы старения и окружающей среды (лентивирусы, дефекты энергетического метаболизма, недостаточность нейротрофических факторов, эксайтотоксичность, митохондриальные дефекты, нейротоксичность микроэлементов и окислительный стресс [20]) и тем меньше влияет генетическая предрасположенность. А чем раньше возраст начала, тем БА будет больше определяться детерминированной мутацией в одном гене [21].
Генетические аспекты БА с ранним началом
БА с ранним началом – заболевание практически с полной генетической детерминированностью, наследуемость составляет от 92 до 100% [22].
У 35–60% пациентов с БА с ранним началом обнаруживается не менее одного родственника первой степени родства с данным заболеванием (т.е. семейная форма БА с ранним началом), причем у 10–15% пациентов с семейной БА с ранним началом отмечается аутосомно-доминантный тип наследования [23]. Аутосомно-доминантная форма БА с ранним началом составляет 0,5% от всех случаев БА [9].
Почти у 50% родственников пациентов с симптомами аутосомно-доминантной формы БА с ранним началом выявляются мутации в следующих генах [24] (табл. 2):
- ген бе...












