Клиническая Нефрология №1 / 2010
Поражение почек при болезни Фабри: возможности радикального улучшения прогноза
Кафедра терапии и профболезней ММА им. И.М. Сеченова, Москва
Представлены особенности диагностики и лечения поражения почек при болезни Фабри.
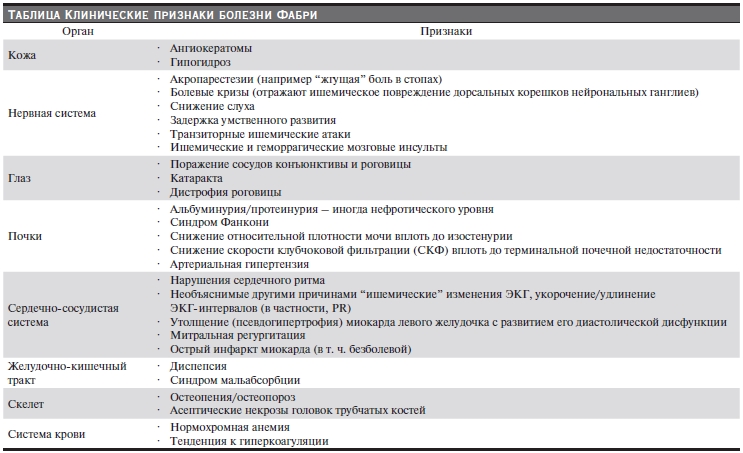
Болезнь Фабри (Андерсона–Фабри) – наследственное заболевание, обусловленное Х-сцепленным дефектом метаболизма сфинголипидов, связанным с недостаточностью a-галактозидазы А [1, 2]. Патогенетическую основу органных поражений при болезни Фабри составляет накопление, в первую очередь в структурах сосудистой стенки (эндотелиоциты, гладкомышечные клетки медии), нейтральных сфинголипидов – церамид-тригезоксида и церамид-дигалактозида, приводящее к тяжелым метаболическим расстройствам и в конечном итоге – к необратимому фиброзу органов-мишеней. Болезнь Фабри может сопровождаться вовлечением практически любых внутренних органов, а также кожи и центральной нервной системы (см. таблицу), что обусловливает обращение этих пациентов к представителям различных клинических специальностей и определенные трудности в своевременной диагностике заболевания. Первыми клинически очевидными проявлениями болезни Фабри, как правило, становятся поражение почек, сердца, а также кожная и неврологическая симптоматика.
Результаты оценки распространенности болезни Фабри среди лиц с соответствующими вариантами органных поражений, которые могут быть обусловлены этим заболеванием, свидетельствуют о том, что оно не является столь редким, как большинство других наследственных заболеваний. Так, по данным скрининга, проведенного в рамках Stroke Prevention in Young Men Study [3] – исследования, включившего молодых (возраст –15–49 лет) мужчин с ишемическим инсультом, в 0,18 % всех его случаев и в 0,65 % случаев ишемического инсульта неустановленного происхождения удается выявить носительство соответствующих генетических детерминант болезни Фабри. Скрининг маркеров болезни Фабри, выполненный у 171 977 новорожденных Тайваня [4], показал, что частота носительства мутации гена, кодирующего a-галактозидазу А, составляет примерно 1 на 1250 новорожденных мальчиков, самым частым вариантом мутации является c.936+919G > A (также обозначаемая как IVS4 + 919G > A), обнаруживаемая у лиц с поздним дебютом поражения сердца. Аналогическое обследование 37104 новорожденных мальчиков, предпринятое в Италии [5], выявило, что у 0,3 % из них имеются как мутации гена, так и снижение активности a-галактозидазы А, что соответствует расчетной частоте заболевания 1 на 3100 новорожденных мальчиков; при этом соотношение позднего и “классического” (дебют болезни в детском возрасте) фенотипов составляет примерно 11 : 1. Эти данные также указывают на то, что большая часть новых случаев болезни Фабри в ближайшие годы будет диагностироваться не педиатрами, а врачами, работающими со взрослыми, – нефрологами, кардиологами, неврологами и дерматологами.
Частоту болезни Фабри неоднократно целенаправленно изучали у пациентов с различными стадиями хронической болезни почек (ХБП), в т. ч. терминальной почечной недостаточностью. По данным US Renal Data System [6], в течение 3 лет (1996–1998) заместительная почечная терапия ежегодно начинает проводиться 11–14 пациентам с нефропатией в рамках болезни Фабри. Анализ европейского регистра ERA-EDTA [7] свидетельствует, что в течение 6 лет (1986–1993) были идентифицированы 83 пациента с терминальной почечной недостаточностью, обусловленной болезнью Фабри, что соответствует 0,0188 % в структуре всех причин необратимого ухудшения функции почек.
Тем не менее можно утверждать, что многие случаи почечного поражения при болезни Фабри остаются нераспознанными и при развитии терминальной почечной недостаточности их учитывают в других нозологических рубриках, что в свою очередь приводит к получению меньших по сравнению с истинными величин распространенности этого заболевания. P. Kotanko et al. (2004) [8] выполнили обследование на наличие болезни Фабри у 2480 пациентов 50 из 55 гемодиализных центров Австрии. На первом этапе снижение активности a-галактозидазы А регистрировали с помощью специального скринингового теста (тест-полоска, при нанесении на которую капли крови происходит изменение цвета). Если тест оказывался положительным, определяли активность a-галактозидазы А в лейкоцитах крови; при ее снижении проводили генетический скрининг мутаций соответствующего гена. У 85 (3,42 %) обследованных был констатирован положительный результат скринингового теста, из них у 4 (3 из них имели установленный диагноз болезни Фабри) активность лейкоцитарной a-галактозидазы А была снижена существенно, еще у 11 – умеренно. Генетический анализ показал, что в целом болезнь Фабри является причиной терминальной почечной недостаточности у 0,161 пациента, находящегося на программном гемодиализе. В ...













{kind=link}