Клиническая Нефрология №2 / 2010
Ранние маркеры диабетической нефропатии
Кафедра эндокринологии Новосибирского государственного медицинского университета, Новосибирск
Диабетическая нефропатия (ДН) занимает одно из ведущих мест в современной структуре причин хронической почечной недостаточности. В статье обсуждаются общепризнанные и перспективные маркеры диабетического поражения почек, в т. ч. его ранних стадий.
Диабетическая нефропатия (ДН) – одно из самых частых, тяжелых и прогностически неблагоприятных осложнений сахарного диабета (СД). В последние годы ДН заняла лидирующие позиции среди причин терминальной почечной недостаточности в индустриально развитых странах. Неудовлетворительные результаты лечения ДН связаны со сложностью патогенеза, длительным бессимптомным течением, поздней диагностикой. Проведенные в разных регионах России выборочные эпидемиологические исследования свидетельствуют, что среди больных СД фактическая распространенность поражения почек превышает регистрируемую в 2–3 раза [1].
В настоящее время не вызывает сомнения, что структурные и функциональные изменения почек появляются в первые годы течения СД. Наиболее ранние признаки почечного поражения включают гиперфильтрацию и клубочковую гипертрофию, далее развиваются утолщение и нарушение структуры базальных мембран, экспансия мезангия клубочков, перигломерулярный склероз, дистрофические и атрофические изменения канальцев, фиброз интерстиция, артерий и артериол [2, 3].
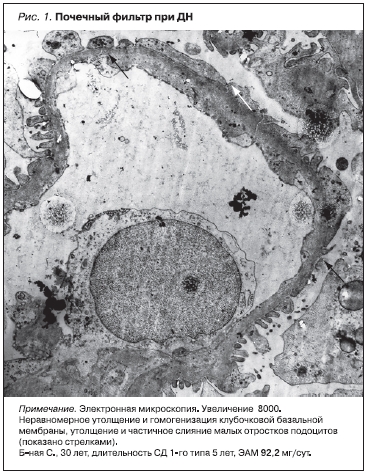
По нашим данным, выраженные изменения гломерулярного барьера, начальные признаки фиброзирования клубочков, интерстиция и сосудов почек у больных СД типа 1 (СД1) обнаруживаются уже на стадии микроальбуминурии (рис. 1, 2). На доклинических этапах ДН при нормальной экскреции альбумина с мочой (ЭАМ) и микроальбуминурии в почечных клубочках начинают накапливаться минорные (IV и VI типы) и интерстициальные (I и III типы) коллагены. В базальных мембранах канальцев и в интерстиции почек возрастает количество коллагена III, IV, в меньшей степени – I и VI типов [4, 5]. Это соответствует экспериментальным данным об активирующем влиянии гипергликемии на синтез коллагенов в клубочковых и канальцевых клетках [6], свидетельствует о раннем начале формирования фиброза почек при СД.
Появление протеинурии у большинства больных СД подтверждает выраженное и необратимое поражение почек [7]. Напротив, более ранние стадии диабетической нефропатии могут подвергаться обратному развитию. В ставшей классической работе P. Fioretto и соавт. показано, что начальные структурные изменения клубочков и интерстиция почек у больных СД1 с микроальбуминурией постепенно (в течение 10 лет) регрессируют при полном устранении гипергликемии после трансплантации поджелудочной железы [8, 9]. Использование современных технологий лечения позволило значительно снизить частоту развития поздних стадий ДН в ряде специализированных центров США, Швеции, Дании [10], а также России [8]. Эти данные указывают на то, что ранняя диагностика и лечение поражения почек имеют принципиальное значение для улучшения прогноза пациентов с ДН. В связи с этим продолжается поиск диагностических маркеров, которые бы отражали наиболее ранние стадии данного осложнения.
Альбуминурия. Выявление микроальбуминурии является стандартным методом диагностики ДН в клинической практике. Нижний порог диапазона микроальбуминурии (30 мг/сут, или 20 мкг/мин) заведомо выше, чем максимальные значения ЭАМ у здоровых лиц. Показано, что у здоровых взрослых альбуминурия обычно не превышает 15 мкг/мин, составляя в среднем около 5 мкг/мин [11]. В свою очередь наличие “высокой нормальной” ЭАМ у больных диабетом является предиктором развития микроальбуминурии [12–14]. Микро- и макроальбуминурия не представляют собой дискретные в клиническом отношении понятия, скорее их можно рассматривать как части континуума: повторные измерения ЭАМ у одних и тех же пациентов могут давать результаты в смежных диапазонах [15].
При оценке результатов определения альбуминурии у больных СД следует учитывать, что к увеличению ЭАМ могут приводить кетоацидоз, высокобелковая диета, чрезмерная физическая нагрузка, хроническая сердечная недостаточность, лихорадка, а также любое сопутствующее заболевание почек. В связи с этим определение микроальбуминурии нужно проводить после устранения кетоацидоза, инфекции мочевыводящих путей, перегрузки белком, на фоне нормальной температуры тела и обычной физической активности пациента. Для определения микроальбуминурии целесообразно использовать суточную порцию мочи. При использовании разовых (утренних) порций мочи необходимо пересчитывать альбуминурию на величину экскретируемого креатинина [15].
Средняя частота развития микроальбуминурии среди больных СД1 составляет 2,5–5,0 % в год. Большинство случаев микроальбуминурии выявляется при длительности заболевания от 5 до 20 лет, при дальнейшем увеличении длительности СД число новых случаев снижается [15]. Кумулятивная частота развития микроальбуминурии при длительности СД1 40 лет составляет 25 % [16]. При СД2 микроальбуминурия может регистрироваться уже в момент диагностики СД, что связано с длительным бессимптомным течением данной формы заболевания, частым наличием сопутствующей артериальной гипертензии и других нефропатий [15].
Распространенность микроальбуминурии при СД варьируется в различных популяциях и зависит от возраста пациентов, длительности диабета, ...













{kind=link}
{kind=link}