Фарматека №2 / 2024
Редкие причины моногенного гипопитуитаризма: обзор литературы
Национальный медицинский исследовательский центр эндокринологии, Москва, Россия
Врожденный гипопитуитаризм – группа редких заболеваний, связанных со снижением или отсутствием секреции одного или более гормонов гипофиза. В настоящее время известно, что развитие заболеваний данной группы связано с мутациями более чем в 30 генах, ответственных за развитие гипоталамо-гипофизарной области. Вариабельность клинической картины обусловлена молекулярно-генетической основой. При этом четкую корреляцию генотипа и фенотипа не всегда возможно определить, поскольку даже у членов одной семьи с одной и той же мутацией клинические проявления могут варьироваться. В данном обзоре рассматриваются известные, а также редко встречающиеся факторы транскрипции и сигнальные молекулы, молекулярные дефекты, которые приводят к развитию изолированного и комбинированного дефицита гипофизарных гормонов у человека.
Введение
Врожденный гипопитуитаризм – редкое заболевание с распространенностью от 1/4000 до 1/10 000 [1]. Врожденный гипопитуитаризм может характеризоваться дефицитом функции одного гормона, чаще всего изолированным дефицитом гормона роста (ИДГР) или дефицитом двух и более гипофизарных гормонов (множественный дефицит гормонов гипофиза – МДГГ). В 5–16% случаев врожденного гипопитуитаризма идентифицируется генетическая основа заболевания [2]. Как правило, мутации возникают в генах, ответственных за онтогенез гипоталамо-гипофизарной области. Однако факт того, что этиология заболевания чаще всего остается неизвестной, позволяет предположить важную роль и других генов в развитии заболевания [3].
Гипофиз – это центральный регулятор, ответственный за контроль роста, метаболизм, развитие, репродукцию и поддержание гомеостаза путем регуляции периферических эндокринных желез в организме. Несмотря на свою главенствующую роль в эндокринной системе организма, гипофиз имеет довольно простую организацию. Ранее ошибочно полагали, что гипофиз представляет собой смесь клеток, но в процессе детального изучения обнаружили, что клетки гипофиза собраны в трехмерные структуры и каждая такая структура выполняет четко определенную роль. Передняя (и выделяемая отдельно в зарубежных источниках как промежуточная) доля гипофиза происходит из оральной эктодермы, в то время как задняя доля – из отростка воронки промежуточного мозга. Развитие гипофиза, а также детерминация и спецификация клеток зависят от экспрессии и взаимодействия сигнальных молекул и транскрипционных факторов в различных, но перекликающихся пространственных и временных паттернах (см. рисунок) [1].
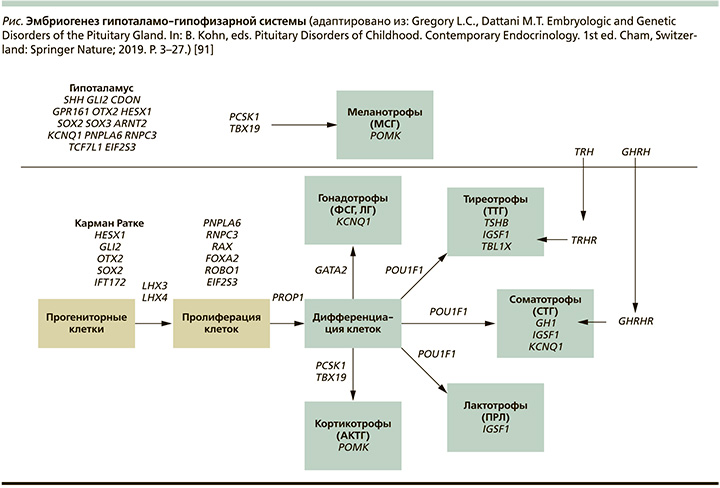
В результате этого взаимодействия формируется пять высокодифференцированных типов клеток, среди которых соматотрофы (вырабатывают СТГ – соматотропный гормон), кортикотрофы (синтезируют АКТГ – адренокортикотропный гормон), тиреотрофы (вырабатывают ТТГ – тиреотропный гормон), гонадотрофы (синтезируют ЛГ – лютеинизирующий и ФСГ – фоликуллостимулирующий гормоны), а также лактотрофы (вырабатывают ПРЛ – пролактин).
С момента картирования первого гена, участвовавшего в развитии гипофиза у человека в 1992 г., существенно расширилось и изменилось понимание генетических основ гипопитуитаризма: за последние 3 десятилетия обнаружены патогенные варианты более 30 генов [4, 5]. Применение современных методов молекулярно-генетического тестирования позволяет идентифицировать все новые гены, участвующие в развитии гипофиза, а также расширять знания о фенотипах, связанных с ранее известными генами.
В таблице представлены гены, мутации в которых ассоциированы с развитием врожденного гипопитуитаризма.
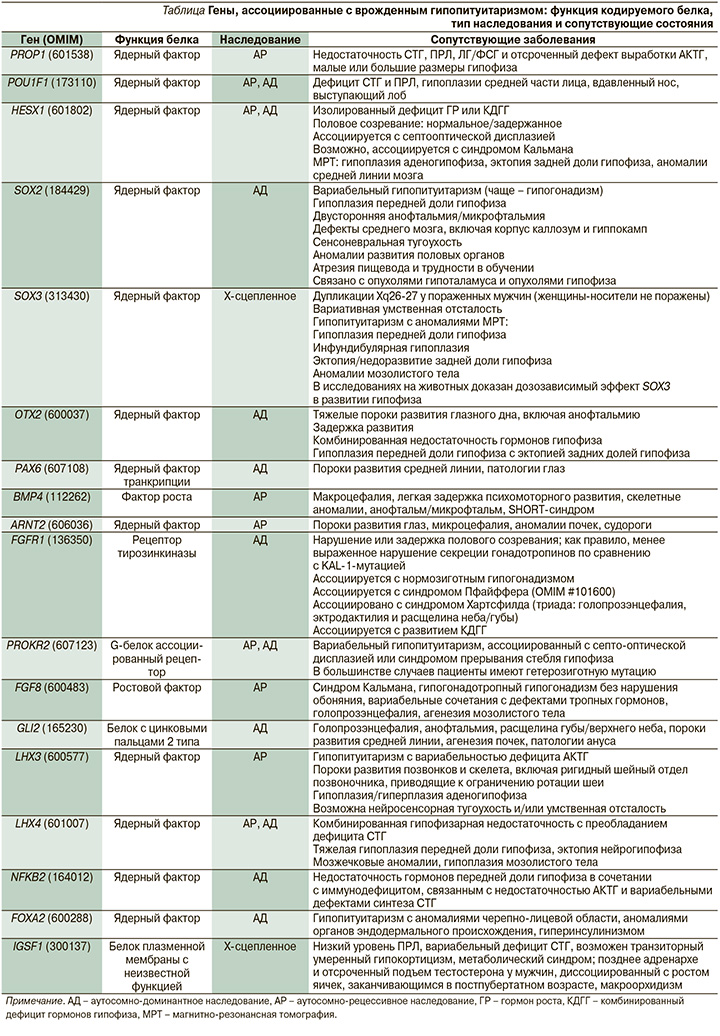
Мутации в генах, участвующих в ранних этапах развития гипоталамо-гипофизарной области (HESX1, LHX3, LHX4, SOX2, SOX3, GLI2, OTX2), связаны со структурными аномалиями не только гипоталамо-гипофизарной оси, но и срединной линии, c внегипофизарными дефектами. Мутации же в генах, экспрессирующихся на более поздних этапах развития (PROP1 и POU1F1), связаны с фенотипом заболевания без экстрагипофизарных дефектов [6].
В неонатальном периоде клинические проявления врожденного гипопитуитаризма неспецифичны и включают апноэ, летаргию, плохой набор массы тела, плохой аппетит; определяются этиологией, возрастом манифестации, а также количеством и типом затронутых тропных гормонов [1, 5, 7].
Задержка роста при дефиците СТГ манифестирует чаще после первого года рождения, реже может проявляться уже в младенчестве. Гиперинсулинемическая тяжелая гипогликемия, связанная с судорожным синдромом, сохраняющаяся более 3 дней и ведущая потенциально к развитию повреждения головного мозга, – наиболее частое проявление дефицита СТГ, а также АКТГ [8]. При этом сочетание дефицита этих тропных гормонов приводит к более тяжелым и частым эпизодам гипогликемии. Дефицит АКТГ характеризуется также гипонатремическими и гипотензивными проявлениями. Признаки дефицита ЛГ в младенчестве – это микропения (SD полового члена менее -2,5 от среднего значения) и крипторхизм, т.к. именно ЛГ отвечает за выработку тестостерона плодом, а также за процесс опускания яичек. Микропения выявляется также при ИДГР, что говорит о большом значении СТГ для роста полового члена во внутриутробном и постнатальном периодах [9, 10].
Неспецифичным, но довольно частым проявлением гипопитуитаризма в неонатальном периоде являются холестатическая желтуха и неонатальный гепатит (что ряд авторов объясняют дефицитом глюкокортикоидов и тиреоидных гормонов) [11]. При этом уровни трансаминаз и γ-глутамилтранспептидазы у пациентов в пределах референса.
Сложн...












