Фарматека №8 (261) / 2013
Роль фармакогенетики в индивидуализации противоопухолевой химиотерапии
Индивидуальный подход к подбору противоопухолевой химиотерапии может быть основан не только на биологических параметрах опухоли, но и на генетически обусловленных особенностях метаболизма цитостатиков, влияющих на их эффективность и токсичность. Эти особенности изучает фармакогенетика, обусловлены же они полиморфизмами (вариациями ДНК) генов, отвечающих за транспорт, метаболизм и цитостатический эффект препаратов. Во многих клиниках фармакогенетическое тестирование используется для индивидуализации химиотерапии иринотеканом, фторурацилом и меркаптопурином, широко обсуждается включение в этот список тамоксифена. В отношении других препаратов роль фармакогенетического тестирования находится в процессе изучения.
Фармакогенетика изучает генетические особенности реакций организма на лекарственные препараты и способна помочь в индивидуальном подборе цитостатиков. Молекулярным субстратом служат вариации ДНК генов, кодирующих белки, которые отвечают за транспорт и метаболизм препаратов или служат их мишенями. В зависимости от распространенности в популяции эти варианты делят на
полиморфизмы (с частотой выше 1 %) и мутации (более редкие) [1–3].
Фармакогенетическое тестирование применяют для выявления генетических вариантов, ведущих к изменению активности белков, которые играют роль в фармакокинетике или фармакогенетике того или иного препарата. Теоретически фармакогенетическое тестирование может позволить скорректировать дозы или даже схему лечения, повысив его эффективность и снизив токсичность.
Ниже будут рассмотрены примеры фармакогенетического тестирования, которые уже вошли в клиническую практику (для индивидуализации химиотерапии иринотеканом, фторпиримидинами и тиопуринами), а также ряд других активно изучаемых клинико-генетических корреляций.
Глюкуронилтрансфераза и иринотекан
Глюкуронилтрансфераза соединяет с глюкуроновой кислотой ряд эндогенных (прежде всего билирубин) и экзогенных субстратов. Существует по крайней мере 17 изоформ этого фермента, объединенных в 2 семейства – UGT1 и UGT2. Ген UGT1A кодирует 9 изоформ подсемейства UGT1, все
они образуются за счет альтернативного сплайсинга. Наибольшее значение имеет изоформа UGT1A1, она отвечает за глюкуронирование билирубина. В гене UGT1A описано более 60 полиморфизмов, многие из которых лежат в основе синдрома Жильбера (легкая форма наследственной непрямой
гипербилирубинемии).
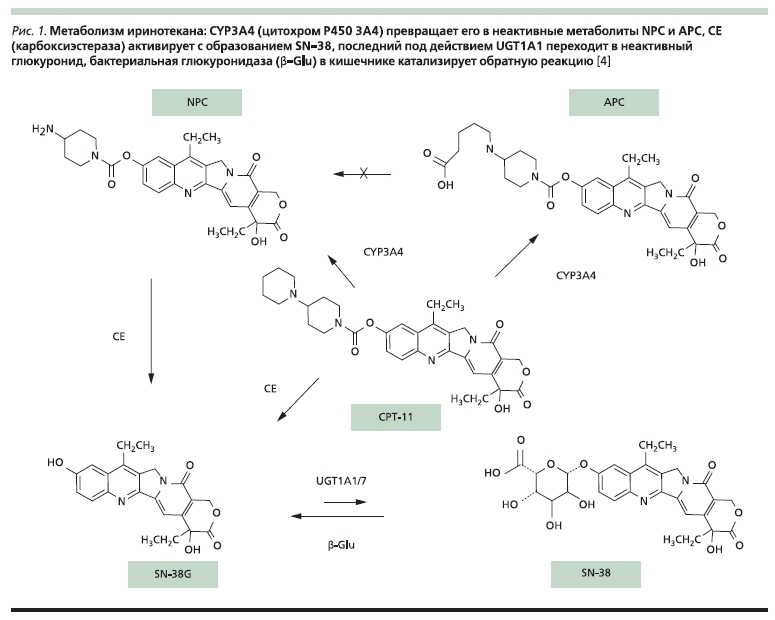
Рисунок 1. Метаболизм иринотекана СНЗА4 (цитохром Р450 3А4) превращает его в неактивные метаболиты NPC и АРС, СЕ (карбоксиэстераза) активирует с образованием SN-38, последний под действием UGT1А1 переходит в неактивный глюкуронид, бактериальная глюкуронидаза (d-Glu) в кишечнике катализирует обратную реакцию [4].
К субстратам UGT1A1А и UGT1A7 относится активный метаболит иринотекана – SN-38 (ингибитор ДНК-
топоизомеразы I, рис. 1). До 15 % населения являются гомозиготами по аллелю UGT1A1*2, сопряженному с увеличением от 6-го до 7-го числа повторов TA (тимин–аденин) в области промотора, что сопровождается снижением экспрессии гена и, соответственно, уменьшением количества белка.
В результате замедляется глюкуронирование SN-38, возрастает риск тяжелой нейтропении и поноса. На фоне высоких доз иринотекана (300–350 мг/м2) риск этих осложнений у гомозигот по аллелю UGT1A1*28 возрастал в 5–10 раз. Таким больным рекомендуется начинать лечение с дозы не выше 250 мг/м2, при хорошей переносимости она может быть повышена в последующих курсах. Для более низких доз корреляция между генотипом и побочными эффектами выражена слабо. Исследования последних лет указывают на более сложную картину: в ряде работ фармакокинетика и токсичность иринотекана лучше коррелировали с другими аллелями (например, UGT1A1*60 и UGT1A7*3); кроме того, по-видимому, имеют значение и другие гены, участвующие в метаболизме препарата и опосредующие его цитотоксичность [4, 5].
Дигидропиримидиндегидрогеназа и фторурацил
Дигидропиримидиндегидрогеназа участвует в катаболизме пиримидиновых оснований, а также инактивирует более 80 % фторпиримидинов (фторурацила и его производных – капецитабина, тегафура, флоксуридина). У 3–5 % европейцев активность фермента снижена, у 0,1–0,2 % он полностью отсутствует. До 50 % случаев недостаточности фермента связано с полиморфизмом IVS14+1G>A (аллель DPYD*2A): он заключается в замене одного нуклеотида, из-за чего нарушается сплайсинг мРНК, выпадает один экзон и образующийся укороченный белок быстро разрушается. Этот полиморфизм встречается у 1–2 % европейцев, с ним связывают около 25 % случаев тяжелой непере-
носимости фторурацила. Примерно так же распространен полиморфизм 2846A>T (т. е. замена аденина на тимин в 2846-м кодоне), вызывающий замену Asp949Val (т. е. аспарагиновой кислоты в 949-м положении на валин) и резкое снижение активности фермента [6, 7].
При выявлении этих полиморфизмов рекомендуется 2-кратное снижение дозы фторурацила и его аналогов или использование альтернативных схем, при носительстве двух вариантных аллелей эти препараты противопоказаны.
Тиопуринметилтрансфераза и меркаптопурин
Тиопуринметилтрансфераза катализирует S-метилирование тиопуринов (меркаптопурина, тиогуанина и азатиоприна), предотвращая образование активных метаболитов – тиогуаниновых нуклеотидов. Ген TPMT имеет полиморфные аллели, при некоторых стабильность фермента резко снижается. Описано 8 аллелей этого гена; наибольшее значение среди них имеют TPMT*2 (238G>C с заменой Ala80Pro),
TPMT*3A [460G>A (Ala154Thr) и 719G>A (Tyr240Cys)] и TPMT*3C (719G>A с заменой Tyr240Cys), причем у европейцев чаще встречается аллель TPMT*2, у афроамериканцев и ази...













{kind=link}