Кардиология №10 / 2016
Синдром Бругада как причина внезапной смерти. Особенности диагностики и клинических проявлений у детей
Научно-исследовательский клинический институт педиатрии им. акад. Ю.Е. Вельтищева ГБОУ ВПО Российский национальный исследовательский медицинский университет им. Н.И. Пирогова Минздрава РФ, Москва
Синдром Бругада — наследственное потенциально аритмогенное заболевание, относящееся к разряду каналопатий, которое проявляется синкопальными состояниями и внезапной смертью у лиц молодого возраста в отсутствие структурного заболевания сердца. В основе заболевания лежит генетически детерминированное нарушение функции ионных каналов кардиомиоцитов (натриевых, калиевых, кальциевых), фенотипически проявляющееся постоянным или транзиторным подъемом сегмента ST на электрокардиограмме, а также высоким риском развития полиморфной желудочковой тахикардии, фибрилляции желудочков и внезапной смерти. У пациентов с высоким риском внезапной смерти единственным доказанным методом профилактики фатальных аритмий и внезапной смерти является имплантация кардиовертера-дефибриллятора.
Синдром Бругада (СБ) — наследственное, генетически детерминированное заболевание, которое характеризуется специфическим элетрокардиографическим паттерном и высоким риском развития потенциально летальных аритмий. В основе заболевания лежит нарушение функции ионных каналов кардиомиоцитов — КМЦ (натриевых, калиевых, кальциевых), которое фенотипически проявляется в виде постоянного или чаще транзиторного подъема сегмента ST на электрокардиограмме (ЭКГ) и обусловливает высокий риск развития полиморфной желудочковой тахикардии (ЖТ), фибрилляции желудочков (ФЖ) и внезапной смерти (ВС) [1]. Как правило, заболевание развивается в отсутствие основополагающей органической патологии сердца, хотя в ряде случаев могут выявляться незначительные структурные изменения.
История вопроса. В 1953 г. H. Osher и L. Wolff опубликовали клинико-электрокардиографическое наблюдение сочетания блокады правой ножки пучка Гиса (БПНПГ) и инфарктоподобного подъема сегмента ST у молодого человека без явных признаков органического заболевания сердца, коронарных артерий, а также электролитных нарушений [2].
В 1989 г. B. Martini и A. Nava описали 6 случаев ФЖ и ВС, у 2 из этих больных была документированы БПНПГ и подъем сегмента ST в правых прекордиальных отведениях [3]. Позже, в 1992 г. Josep и Pedro Brugada, проанализировав данные 8 пациентов с рецидивирующими синкопальными состояниями (СС), эпизодами ВС вследствие ФЖ, а также БПНПГ и транзиторным подъемом сегмента ST в правых прекордиальных отведениях, предложили считать это новым клинико-электрокардиографическим синдромом, впоследствии названным по фамилии авторов [4]. Последующие многочисленные исследования с проведением ангиографии, компьютерной и магнитно-резонансной томографии сердца, биопсии миокарда не выявили у таких больных клинически значимых структурных заболеваний сердца и сосудов.
 Синдром внезапной необъяснимой смерти встречается во всем мире, но наиболее часто (до 43 случаев на 100 000 человек в год) — в странах Восточной Азии, где этим термином обозначаются случаи необъяснимой смерти здоровых молодых мужчин в возрасте от 20 до 40 лет, возникающие, как правило, во сне или в покое, а также часто после обильного приема пищи [5, 6]. На Филиппинах этот синдром впервые был описан в 1917 г. под оригинальным названием Ban-gun-gu («поднялся и застонал» — на местном языке). В Японии синдром впервые идентифицирован как болезнь pokkuri («внезапная смерть») в 1959 г., в Тайланде и Лаосе — как laitai («смерть во сне»), на Гаваях — «dream disease». В этих краях существовало поверье о том, что, если человека разбудить в этот момент, можно избежать смерти [5—7].
Синдром внезапной необъяснимой смерти встречается во всем мире, но наиболее часто (до 43 случаев на 100 000 человек в год) — в странах Восточной Азии, где этим термином обозначаются случаи необъяснимой смерти здоровых молодых мужчин в возрасте от 20 до 40 лет, возникающие, как правило, во сне или в покое, а также часто после обильного приема пищи [5, 6]. На Филиппинах этот синдром впервые был описан в 1917 г. под оригинальным названием Ban-gun-gu («поднялся и застонал» — на местном языке). В Японии синдром впервые идентифицирован как болезнь pokkuri («внезапная смерть») в 1959 г., в Тайланде и Лаосе — как laitai («смерть во сне»), на Гаваях — «dream disease». В этих краях существовало поверье о том, что, если человека разбудить в этот момент, можно избежать смерти [5—7].
Как самостоятельная нозологическая форма синдром внезапной необъяснимой смерти во сне был впервые идентифицирован в США в начале 80-х годов XX века у переселенцев из Восточной Азии [6]. В 2002 г. М. Vatta и соавт. показано, что «sudden unexpected nocturnal death syndrome» (SUNDS), описанный у жителей Восточной Азии, и СБ, описанный у европейцев, – генетически и патофизиологически идентичные состояния [8].
Демографические данные. СБ является редким заболеванием. Динамичность электрокардиографических признаков и неспецифичность клинических проявлений не позволяют достоверно оценить распространенность синдрома в общей популяции. Имеющиеся эпидемиологические данные показывают низкую распространенность СБ в общей популяции. Так, в европейских странах синдром встречается с частотой от 1,1 до 10 на 100 000 человек в возрасте старше 21 года [9—11]. Однако ряд экспертов считают, что истинная распространенность СБ намного выше и составляет от 1:2000 до 1:5000 [11].
Распространенность синдрома у детей неизвестна. Следует отметить, что 3 из первых 8 пациентов, впервые описанных братьями Бругада, на момент начала СБ было от 2 до 8 лет.
Клинические проявления СБ варьируют от длительного, в ряде случаев на протяжении всей жизни, бессимптомного течения до ФЖ и ВС в качестве первого и единственного симптома. Заболевание может манифестировать в любом возрасте: симптомы, обусловленные возникновением потенциально фатальных тахиаритмий, описаны у пациентов с СБ в возрасте от 2 дней до 84 лет [12]. Пик клинических проявлений в виде эпизодов ФЖ и ВС приходится на возраст 41±15 лет. В 75—80% случаев поражаются мужчины. При этом фатальные аритмии, включая ФЖ, в 5 раз чаще регистрируются у лиц мужского пола. В 20—50% случаев семейный анамнез отягощен по ВС [11, 12].
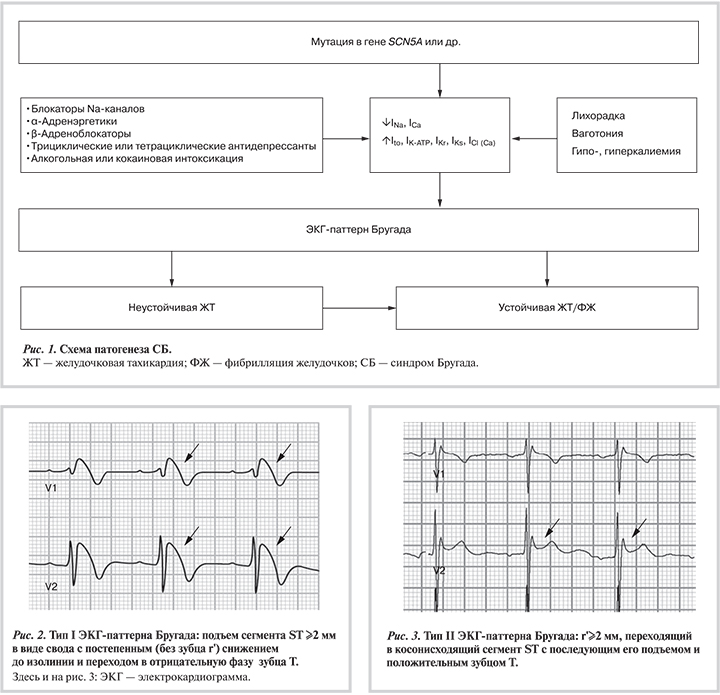
В общей популяции частота ВС варьирует от 1,3 до 4,3 случая на 100 000 человек в год [12]. На ее долю приходится почти 19% всех ВС у детей в возрасте от 1 до 13 лет. СБ служит причиной около 4% всех случаев ВС и по крайней мере до 20% случаев всех ВС в отсутствие структурной патологии сердца [12—14]. В настоящее время доказано, что СБ является также одной из причин ВС у детей, в частности, умерших от синдрома ВС младенцев [15].
ЭКГ-пат...












