Клиническая Нефрология №2-3 / 2015
Влияние генетической тромбофилии на тяжесть течения гемолитико-уремического синдрома у детей
1 ГБУЗ «Детская клиническая больница Святого Владимира», Москва; 2 ГБОУ ВПО «Московский государственный медико-стоматологический университет им. А.И. Евдокимова»; 3 ГБОУ ВПО «Первый Московский государственный медицинский университет им. И.М. Сеченова» МЗ РФ; 4 ГБУЗ МО «Московский областной научно-исследовательский клинический институт им. М.Ф. Владимирского»
Цель. Изучить влияние генетически обусловленной тромбофилии на тяжесть течения гемолитико-уремического синдрома (ГУС) у детей.
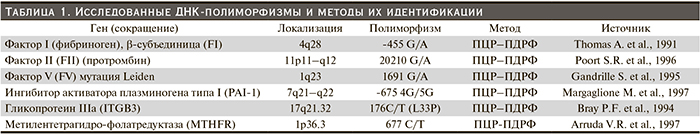
Материал и методы. Обследованы 47 пациентов с типичной формой ГУС (средний возраст – 19,3±2,1 месяца). Методом ПЦР-масс-спектрометрии и ПЦР-рестрикционного анализа исследованы генетические маркеры тромбофилии: алелльный полиморфизм генов фибриногена (FGB), протромбина (PTG), метилентетрагидрофолатредуктазы (MTHFR), ингибитора активатора плазминогена (PAI-1), фактора V Лейден (F5), тромбоцитарного рецептора фибриногена (ITGB3).Проведена оценка влияния полиморфизма изучаемых генов системы свертывания на клинические проявления ГУC (длительность анурии, олигурии, гиперазотемии, анемии, диализной терапии, выраженность тромбоцитопении, протеинурии в остром периоде и при выписке).
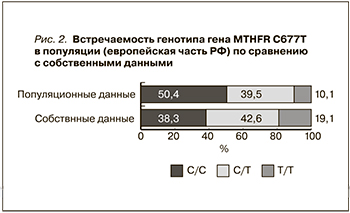
Результаты. У детей с ГУС по сравнению с распределением в популяции в 2 раза чаше встречался гомозиготный генотип генов MTHFR и ITGB; существенно чаще – гетерозиготы генов FGB и PAI-1. Дети с мутантным вариантом полиморфизма гена MTHFR С677Т представляют группу с более тяжелым течением ГУС. У пациентов с «протромбогенным генотипом» гена FGB также отмечены более длительная анемия, анурия, гиперазотемия, диализная терапия. Наличие гетеро- и гомозиготных генотипов гена PAI-1 4G(675)5G и ITGB3 С176Т влияло на длительность анурии, диализной поддержки и периода восстановления почечных функций.
Заключение. Выраженность клинических проявлений ГУС определяется «протромбогенным генотипом» генов MTHFR, ITGB, FGB и PAI-1.
Введение
Исследования последних лет позволили установить связь между развитием тромбозов различной локализации и генетически обусловленной тромбофилией в отсутствие традиционных факторов риска. Вероятность формирования гиперкоагуляционного состояния возрастает с числом выявленных «прокоагулянтных» генотипов. При ГУС развитие микроциркуляторных тромбозов служит основным клиническим проявлением заболевания и поражение почек, как правило, сочетается с тромботической микроангиопатией сосудов головного мозга, легких, кишечника, печени, сердца, приводя к развитию полиорганной недостаточности с нарушением витальных функций. Роль тромбофилии в развитии патологии человека хорошо изучена в акушерской практике, неврологии, кардиологии, нефрологии [1–3]. Возможность поражения сосудов микроциркуляторного русла с развитием тромботической микроангиопатии была четко установлена при антифосфолипидном синдроме. Недавние исследования продемонстрировали развитие нефропатии у больных с «протромбогенными генотипами» генов MTHFR C677T, PAI-1 -675 4G/5G, FGB-455 G/A, ITGB3 L33P и доказали, что наличие мультигенной тромбофилии способствует более быстрому прогрессированию гломерулонефритов [1]. До настоящего времени роль наследственной тромбофилии в развитии и прогрессировании тромботической микроангиопатии у пациентов с ГУС, при котором почки являются главным органом-мишенью для тромбоза сосудов малого калибра, до сих пор не изучена. Комплексное изучение ассоциаций генетических маркеров тромбофилии, клинических особенностей ГУС может расширить представление о прогностических факторах, отдельных патогенетических механизмах, особенностях течения и подходах к лечению данного синдрома.
 Целью настоящего исследования стало изучение влияния генетически обусловленной тромбофилии на характер течения ГУС у детей.
Целью настоящего исследования стало изучение влияния генетически обусловленной тромбофилии на характер течения ГУС у детей.
Материал и методы
В исследование включены 47 пациентов с типичной формой ГУС в возрасте от 5 до 62 месяцев (19,3±2,1 месяца), из которых в возрасте от года до 3 лет были 59,6% (n=28); до года – 27,7% (n=13) и старше 3 лет – 12,8% (n=6) детей. Среди пациентов преобладали мальчики (66%, n=31).
Обследование включило стандартный набор клинико-лабораторных тестов, применяемых в отношении всех больных острой почечной недостаточностью. Всем больным исследовался аллельный полиморфизм шести различных генов системы свертывания крови: метилентетрагидрофолатредуктаза (MTHFR С677Т); ген протромбина (PTGG20210A); V фактора свертывания крови (FVLeidenG1691A); фибриногена (FGBG455A); тромбоцитарного рецептора фибриногена ITGB3 T176CL33P и ингибитор активатора плазминогена (PAI-1 4G/5G 675).
При анализе аллельных полиморфизмов генов выявлен нормальный генотип – «дикий», когда отсутствовали замены, гетерозиготный – при замене 1-го аллеля, либо гомозиготный – при замене 2 аллелей.
Методика исследования ДНК-полиморфизмов представлена межклинической лабораторией молекулярных методов диагностики Первого МГМУ им. И.М. Сеченова (зав. лаб. к.м.н. Е.М. Пальцева).
Для определения полиморфных аллелей генов MTHFR С677Т, PTGG20210A, FVLeidenG1691A, FGBG455A, ITGB3 T176CL33P использован метод полиморфизма длины рестриктивных фрагментов (ПЦР-ПДРФ); а гена PAI-1 4G/5G 675 – метод однонитевого конформационного полиморфизма ДНК (ПЦР- SSCP) (табл. 1).
Статистический анализ проведен на персональном компьютере с использованием статистического пакета SPSS-21.0. Посредством сравнения средних величин с использованием критерия Стьюдента проведена оценка параметрических данных. Правильность распределения событий в группах оценивалась с помощью сравнения с нулевой гипотезой. Во всех группах распределение было правильным и значимо не отличалось от нулевой гипотезы. При р<0,05 разница в группах считалась достоверной.
Результаты
Среди 47 пациентов с ГУС 18 (38,3%) имели преобладающий в популяции генотип (G/G) («дикий») гена β-цепи фибриногена (FGBG(455)A), 26 (55,3%) пациентов обладали гетерозиготным генотипом (G/A) и лишь 3 (6,4%) детей оказались носителями гомозиготного (A/A-«мутантного») генотипа (рис. 1).
 Сравнение длительности анурии у больных с различными генотипами гена фибриногена показало, что у пациентов с носительством генотипа A/A значение этого показателя достоверно превосходило таковое у больных, имеющих «дикий» и гетерозиготный генотипы (р=0,007 и р=0,034). Длительность анемии у детей с генотипами G/G и G/A была соизмеримой и почти в 2 раза меньшей по сравнению с носителями генотипа А/А. Дети с мутантным генотипом А/А имели более выраженную тромбоцитопению по сравнению с носителями генотипов G/G и G/A (табл. 2).
Сравнение длительности анурии у больных с различными генотипами гена фибриногена показало, что у пациентов с носительством генотипа A/A значение этого показателя достоверно превосходило таковое у больных, имеющих «дикий» и гетерозиготный генотипы (р=0,007 и р=0,034). Длительность анемии у детей с генотипами G/G и G/A была соизмеримой и почти в 2 раза меньшей по сравнению с носителями генотипа А/А. Дети с мутантным генотипом А/А имели более выраженную тромбоцитопению по сравнению с носителями генотипов G/G и G/A (табл. 2).
У больных с генотипом А/А средняя продолжительность гиперазотемии почти вдвое превышала таковую при двух других генотипах, составив 70,3±26,3 ...












