Фарматека №4 / 2019
Все ли мы знаем о возможностях метформина в лечении сахарного диабета 2 типа? Какая из его комбинаций наиболее изучена, эффективна и доступна для пациента в реальной практике врача-эндокринолога?
Московский государственный медико-стоматологический университет им. А.И. Евдокимова, Москва, Россия
Заболеваемость сахарным диабетом 2 типа (СД2) неуклонно растет во всем мире, а эффективность контроля гликемии при этом заболевании все еще недостаточна. Обсуждаются патогенетические механизмы СД2. Подчеркивается важная роль инсулинорезистентности как ключевого звена развития СД2. Детально рассматриваются молекулярные механизмы действия метформина, анализируются его гликемические и внегликемические эффекты, место препарата в современной терапии СД2.
Для цитирования: Бирюкова Е.В., Соловьева И.В. Все ли мы знаем о возможностях метформина в лечении сахарного диабета 2 типа? Какая из его комбинаций наиболее изучена, эффективна и доступна для пациента в реальной практике врача-эндокринолога? Фарматека. 2019;26(4):111–21. DOI: https://dx.doi.org/10.18565/pharmateca.2019.4.111-121
Введение
В России, как и во многих других странах, сахарный диабет 2 типа (СД2) является социально значимым заболеванием. Распространенность СД2 крайне высока и увеличивается с каждым годом, однако эффективность контроля гликемии при этом заболевании все еще недостаточна. Этим обусловлен рост частоты микро- и макрососудистых осложнений СД2, определяющих прогноз жизни пациента. Согласно клиническому исследованию PANORAMA (кросс-секционный анализ данных по гликемическому контролю, собранных у 5817 пациентов с СД2), в Европе большая доля пациентов с СД2 имеет уровень гликированного гемоглобина (HbA1c) ≥7% [1] (рис. 1).
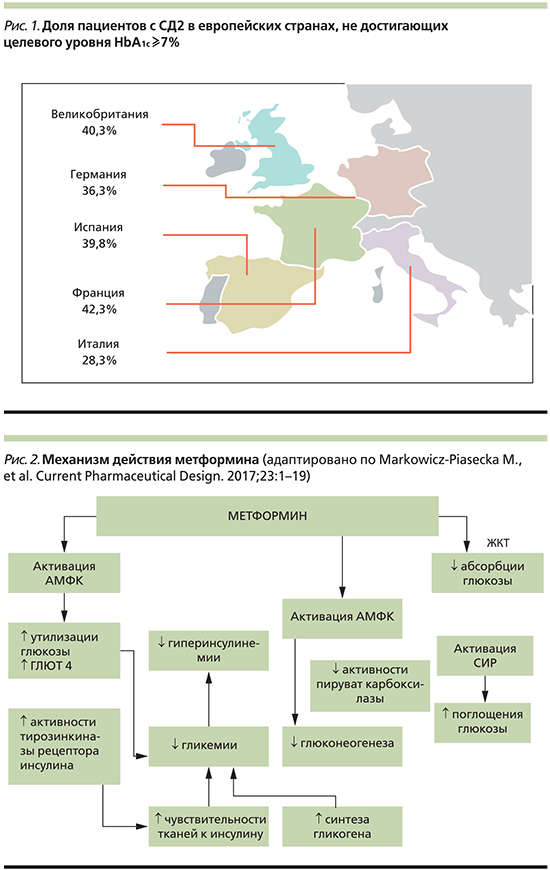
Масштабность проблемы еще более значительна, если принять во внимание тот факт, что наряду с официально зарегистрированными случаями СД2 у значительной части населения диагноз не установлен и имеет место нарушенная толерантность к глюкозе (НТГ) или нарушение гликемии натощак [2]. Согласно исследованию NATION с использованием анкетирования и скринингового определения HbA1c, почти одна пятая населения России находится в стадии предиабета (19,26%) [3]. Высокий риск развития сердечно-сосудистых катастроф имеют не только пациенты с СД2, но и лица с предиабетом. Согласно данным исследования E. Selvin et al. (n=15 792), уровень HbA1c в диапазоне 5,5–6,0% ассоциировался с увеличением риска развития сердечно-сосудистых заболеваний (ССЗ) на 30%, инсульта на 20%, смерти по любой причине на 20%; повышение HbA1c до 6,0–6,5% приводило к увеличению ССЗ на 90%, инсульта в 2 раза, смерти по любой причине на 60% [4]. В исследовании Multi-Ethnic Study of Atherosclerosis (MESA), включившем 10-летний период наблюдения за 6000 лицами, установлено, что предиабет сопровождается трехкратным повышением риска инфаркта миокарда по сравнению с лицами с нормальной толерантностью к глюкозе [5].
Метаболические нарушения, приводящие к развитию микро- и макрососудистых осложнений, возникают задолго до клинического дебюта СД2 и к моменту диагностики недуга приводят к необратимым сосудистым и неврологическим нарушениям. Считается, что первым метаболическим дефектом является инсулинорезистентность (ИР), развитие которой определяет взаимовлияние генетических и приобретенных факторов [6, 7]. В механизмах ее формирования важную роль играют такие нарушения, как липотоксичность, воспаление, глюкотоксичность, митохондриальная дисфункция [8]. Степень ИР и заболеваемость СД2 наиболее выражены среди лиц с абдоминальным ожирением. В клинической практике для его диагностики используется измерение окружности талии [9]. О наличии абдоминального ожирения свидетельствует увеличение окружности талии более 94 см у мужчин и более 80 см у женщин [20].
В условиях тканевой ИР увеличивается продукция инсулина печенью, снижается утилизация глюкозы периферическими тканями. Существует прямая связь между увеличением продукции глюкозы печенью и гипергликемией натощак [7, 10]. Печень играет ключевую роль в регуляции гомеостаза глюкозы, которая выделяется из этого органа в результате гликолиза и глюконеогенеза (1,8–2,2 мг/кг/мин) [9]. Вклад этих метаболических процессов в поддержание углеводного гомеостаза может меняться в разное время суток. В частности, в ночное время около 75% глюкозы печеночного происхождения образуется путем гликогенолиза, а оставшаяся часть – путем глюконеогенеза. Однако по мере удлинения периода ночного голодания глюконеогенез все более преобладает над гликогенолизом [11].
Компенсаторная гиперинсулинемия (ГИ), развивающаяся в условиях снижения чувствительности тканей к действию инсулина, усиливает поглощение глюкозы периферическими тканями, а также уменьшает гепатическую продукцию глюкозы, что определенное время поддерживает нормальное содержание глюкозы крови [6]. Компенсаторная ГИ сопровождается гиперплазией, увеличением количества β-клеток и повышением экспрессии генов ключевых ферментов, участвующих в метаболизме глюкозы. Ослабление подавления гепатической продукции глюкозы после приема пищи играет значимую роль в ухудшении гомеостаза глюкозы в постпрандиальный период [12]. Свой вклад в данный процесс вносят изменения в секреторной функции β-клеток на фоне постоянной стимуляция в сочетании с генетическими дефектами их функции и воздействием липотоксичности. Кроме того, избыток свободных жирных кислот (СЖК) стимулирует печеночный глюконеогенез, подавляет транспорт глюкозы в мыщцы [13].
Истощение ресурсов β-клеток (дефицит ранней фазы секреции инсулина) способствует развитию НТГ, а затем и клиническому СД2. Сама по себе хроническая гипергликемия снижает способность β-клеток секретировать необходимое количество инсулина в ответ на различные стимулы (феномен глюкотоксичности) [14, 15].
ГИ и ИР – независимые факторы риска развития ишемической болезни сердца (ИБС). Для предупреждения сердечно-сосудистой смертности необходимо раннее начало лечения нарушений углеводного обмена (на стадии предиабета) [16].
Выбор ИР в качестве мишени лечения нарушений углеводного обмена позволяет улучшать чувствительность...












